
Abstract
Primary hepatocellular carcinoma (HCC) is currently the fifth most common malignancy and the third most common cause of cancer mortality worldwide. Because of its high prevalence in developing nations, there have been numerous efforts made in the molecular characterization of primary HCC. However, a better understanding into the pathology of HCC required software-assisted network modeling and analysis. In this paper, the author presented his first attempt in exploring the biological implication of gene co-expression in HCC using actor-semiotic network modeling and analysis. The network was first constructed by integrating inter-actor relationships, e.g. gene co-expression, microRNA-to-gene, and protein interactions, with semiotic relationships, e.g. gene-to-Gene Ontology Process. Topological features that are highly discriminative of the HCC phenotype were identified by visual inspection. Finally, the author devised a graph signature-based analysis method to supplement the network exploration.
Keywords
Introduction
Primary hepatocellular carcinoma (HCC) is the fifth most common malignancy and the third most common cause of cancer mortality worldwide with one million new cases diagnosed annually. Its prevalence is much higher in developing nations than in industrialized nations. At present, 80% of the HCC cases came from the East Asia and the sub-Saharan Africa with China accounting for nearly 55% of them [1. For this reason, there have been numerous efforts made in the molecular characterization of primary HCC. As a result, there is a rich repository of genomic and proteomic data available for public access [2. To uncover the biology hidden within such a large volume of data will require software-assisted network modeling and analysis (reviewed in [3). In recent years, attempts to characterize disease phenotypes by integrative network modeling and analysis have been made. For example, Tuck et al. [4 retrieved the human gene regulatory network from the TRANSFAC® database and integrated it with the transcription factor-to-target genes co-expression network derived from multiple microarrays. They then demonstrated that node degree measures are a feasible discriminator of oncology types. Chuang et al. [5 characterized proteomic sub-networks as the biomarkers for discriminating between metastatic and non-metastatic breast cancer. They demonstrated that the protein sub-networks identified are highly discriminative of metastasis and some of the genes underscored by statistical inference methods were found to be member nodes of those sub-networks. These studies demonstrated the effectiveness of network modeling and analysis.
This paper presents the author's first attempt in exploring the biological implication of gene co-expression in HCC using actor-semiotic network modeling. The rationale was that a complex network requires context or metadata to be comprehensible. Without which, no human user would be able to unpack the information content within, let alone making biological deductions. The proposed
Because the topology of an actor-semiotic network is determined by the combination of inter-actor and semiotic relationships, there should be visually identifiable topological features that are highly discriminative of the HCC phenotype. To achieve this, the author employed visual inspection and, in addition, a graph signature-based analysis method to supplement network exploration. This method first summarized the local topology of every node in the network as a signature vector and then projected the vectors onto a two-dimensional scatterplot for further exploration.
Topological Analysis of the Actor-Semiotic Network
Visual Analysis
Using NetMap Decision Director™, an actor-semiotic network
A smaller network

Exploring the actor-semiotic network of HCC. (1) The network

Network topology of
From the largest cluster

Network topology of
The eccentricity and radiality centralities were found to give identical rankings. The same was also observed with the HITS-Authority and HITS-Hub centralities. Therefore the radiality and the HITS-Hub centralities were excluded from the signature vector of each node. After the signature vectors were computed and scaled, the scatterplot shows that there are two clusters of nodes, each representing a different range of signature vectors (Fig. 4). Nodes within the emergent groups were found in the upper cluster and liaison nodes were found in the lower cluster.

Scatterplot generated by projecting graph signatures of
The six nodes at the left-extremity (x-range = [–1661.93, –1617.66]; y-range = [–74.14, 61.57]; Fig. 4) of the lower cluster have signatures that contained the top 5% ranking in closeness, current-flow betweenness, current-flow closeness, and shortest-path betweenness centralities. Three of these nodes
The three nodes at the bottom corner of the lower cluster (x-range = [119.32, 173.93]; y-range = [–1570.32, –1597.41]; Fig. 4) have signatures that contain the bottom 10% ranking in closeness, current-flow closeness, eccentricity, and HITS-authority centralities, and the top 10% ranking in degree, current-flow betweenness, and shortest-path betweenness centralities. They are
In summary, the ranking of all centralities decreases as one moves to the right end of the x-axis in the scatterplot. On the other hand, the node ranking on degree, current-flow betweenness, and shortest-path betweenness centralities increase as one moves to the lower end of the y-axis but at the same time, the rankings on closeness, current-flow closeness, eccentricity, and HITS-authority centralities decrease. The rank score of those nodes mentioned in this paper are tabulated in Table 1.
Node centrality ranking of actor and semiotic nodes in

The actor nodes are listed in the alphabetical order of their gene symbols. The semiotic nodes are listed in the alphanumerical order of the Gene Ontology ID. The rank score for each centrality type ranges from 1 to 1372. A lower rank score means a higher node ranking for a particular centrality type.
Based on the visual exploration of network
de-Synchronized Cell Cycle Phases
The semiotic nodes in emergent group 3 indicated that it contains exclusively cell cycle genes (Fig. 3). Their co-expression was found only in HCC and could be a result of replication stress. Within this emergent group,
Abnormal Angiogenesis
Disrupted Nuclear Transport
Recent findings revealed that many growth factors, e.g.
Discussion
Strength and Limitations of Network Analysis
Network analytics is very suited to biomedical research where high informational granularity and connectivity between objects are required for knowledge inference. However, the scale of the network often presents a cognitive challenge to the analyst. This limitation is partly moderated with the use of NetMap™ which allows the analyst to downsize a large network (|
Biological Implication of Node Centrality
There have been several views on how node centralities signify the biological essentiality of a protein. The first view took degree centrality as the primary indicator of biological essentiality because high degree protein nodes, also known as hubs, are essential for maintaining network connectivity [27. The second view argued that shortest-path betweenness centrality is a better indicator of essentiality [28. This view suggested that bottleneck proteins linked to multiple protein hubs are also biologically essential. The positive correlation between node degree and biological essentiality has been confirmed recently [29, 30] but the original rationale has been challenged [30. Zotenko et al.'s [30 proposition was that the hubs are essential because they form modules in which the member proteins are highly inter-connected and share a common biological function. They named the module as Essential Complex Biological Module (ECOBIM) because it is enriched in essential proteins. Furthermore, the authors demonstrated that current flow betweenness and shortest-path betweenness centralities are better indicators of connectivity, thus supporting the second view. So far, the above hypotheses were deduced from the yeast protein interaction network [27, 28, 30] and the human disease gene network [29 but how do they contribute to the current understanding of cancer biology?
The first view seemed to agree with the recent suggestion that it could take three mutated genes or fewer to induce early stage malignancy [31 since some well studied cancer genes, e.g.
In the network
Thus far, none of the microRNA nodes found in
Conclusion
The use of actor-semiotic network modeling and analysis does provide insight into the pathology of HCC. Although the inclusion of semiotic nodes increases the size of a network, they are useful for identifying discrete clusters or emergent groups that serve a particular biological process or a set of inter-related molecular functions. The provisions of network decomposition and sub-network extraction functionalities by NetMap™ facilitated the ‘top down’ exploration of a large graph. The use of graph signatures further facilitated network exploration by providing a summary of node topologies in a form of a scatterplot.
Methods
Data Sources
Gene expression data
The gene co-expression profiles of HCC and normal hepatocytes were obtained from Gamberoni et al. [36 which was derived from the original dataset published by Chen et al. [37. A set of co-expressed genes from each sample set (normal hepatocyte or HCC) was extracted based on their Pearson's correlation coefficients (
MicroRNA expression data
The microRNA expression data of HCC and adjacent normal hepatocytes was published by Murakami et al. [15. The predicted microRNA target genes were curated from three publications [39–41].
Gene Ontology
The three categories of GO–-Component, Process, and Function, were obtained from the Gene Ontology Consortium [42.
Human proteome data
The canonical human proteomic interaction data was obtained from the BioGrid version 2.0.36 [43. This was integrated with the Hepatitis B-to-human proteomic interaction data obtained from the NCBI Gene RIF.
Data-to-Network Mapping
A relational database was constructed for storing the above datasets. Data for the edges were stored in four tables with each storing data of a specific edge type. The mapping of data to nodes and edges was done with the use of NetMap Decision Director™. The actor nodes are
Network Visualization and Interactivity
The visualization for the networks described in this paper was generated with the use of NetMap™. The software also allows the analyst to (1) decompose a large graph into a set of discrete clusters; (2) extract the largest cluster and identify its largest connected component; (3) decompose the largest connected component to inter-connecting emergent groups; (4) navigate from point-to-point within each network; and (5) search nodes by Gene Symbols or GO identifiers.
Emergent Groups
The identification of emergent groups was completed by a proprietary pattern recognition algorithm embedded in NetMap™. These groups are so named because they
Given an emergent group
|
Each node ν ∊
Under these criteria,
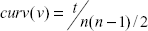
where
Node centralities are metrics for measuring the connectivity pattern of a node in relation to its surrounding neighbours. In this study, nine types of node centralities were calculated using CentiBiN [46. They are closeness, current-flow betweenness, current flow closeness, degree, eccentricity, HITS-authority, HITS-hub, radiality, and shortest-path betweenness centralities. The rationale behind each measure can be found in [47.
Signature Vectors
After computing each node centrality type, the nodes were ranked in the descending order of their centrality values. The node with the highest value for, say degree centrality, would be assigned a rank score of 1. Hence the lower is the rank score, the higher is the node ranking for a certain centrality type. This step generated a column vector
Software Availability
The NetMap Analytics™ software suite which includes NetMap Decision Director™ and NetMap™ is available from NetMap Analytics Proprietary Limited, Sydney, Australia (http://www.netmapanalytics.com.au) under an academic license.
Disclosure
The author reports no conflicts of interest. He did not receive any monetary reward from the NetMap Analytics Proprietary Limited for conducting this research.
Footnotes
Acknowledgments
The author acknowledged Georgina Lakeland from the NetMap Analytics Proprietary Limited for providing technical support. He was also grateful to Dr. Bing Yu from the Faculty of Medicine, the University of Sydney, for his helpful comments on this manuscript.