
Abstract
Gastric polyps are frequently encountered on endoscopic examinations. While many of these represent true epithelial lesions, some of the polyps may result from underlying stromal or lymphoid proliferations or even heterotopic tissue. Histologic examination is essential for accurate typing of the polyps to predict malignant potential and underlying possible genetic abnormalities. The focus of this review is on gastric hamartomatous polyps, which are relatively rare and diagnostically challenging. Though most of the gastric hamartomatous polyps are benign, certain types are associated with increased malignant potential. These include certain polyps associated with specific genetic familial polyposis syndromes and gastric inverted hamartomatous polyps. Identification of these polyps can result in the prevention or early diagnosis of gastric carcinoma and also help in the identification of family members with polyposis syndromes. The aim of this review is to categorize gastric hamartomatous polyps and aid in the identification of high-risk categories.
Introduction
Gastric polyps are encountered in approximately 1%-6.35% of endoscopies.1–3 Gastric polyps are rarely symptomatic and are usually discovered incidentally on endoscopy. 1 The larger gastric polyps may present with bleeding, anemia, obstructive symptoms, and pain. 4 The most common types of gastric polyps are fundic gland polyps (FGPs), hyperplastic polyps, and adenomas.3,5,6 Gastric neuroendocrine tumors (carcinoids), infiltrates (such as xanthomas and lymphoproliferative neoplasms), mesenchymal proliferations (such as leiomyomas, gastrointestinal stromal tumors, and inflammatory fibroid polyps), and others can also be present as polyps.2–4 Gastric hamartomatous polyps are uncommon and comprise about 1% of all the stomach polyps. 7 Most gastric polyps are difficult to characterize on the basis of endoscopic appearance alone and need histologic characterization for assessment of malignant potential. 4 The aim of this review is to characterize the clinical and pathological features of gastric hamartomatous polyps.
Hamartomatous polyps are characterized by disorganized growth of tissue indigenous to the site. 4 They can be solitary or syndromic. 8 The syndromes commonly associated with gastric hamartomatous polyps are Peutz–Jeghers syndrome (PJS), juvenile polyposis, and phosphatase and tensin homolog (PTEN) hamartoma syndrome (PTHS). In the solitary group, solitary juvenile polyps and Peutz–Jeghers type polyps have been reported.8,9 Also described infrequently are inverted gastric hamartomatous polyps, which are submucosal lesions characterized by inverted growth pattern of gastric glands. 7 It is important to distinguish sporadic or solitary polyps from syndromic polyps as the sporadic or solitary polyps generally have a relatively benign course, while those associated with a syndrome have a higher lifetime malignancy risk (though specific gastric cancer risk data are not available). 8 An exception in the sporadic group is gastric inverted hamartomatous polyps (GIHPs), which have a 20% risk of being associated with adenocarcinoma.7,10
PJS Polyps
PJS is an autosomal dominant inherited disorder, characterized by a germline mutation in STK11 mutation. 11 In 1921, Peutz reported a Dutch family with gastrointestinal polyposis and distinctive pigmentation of the skin and mucous membranes and highlighted the inherited nature of the syndrome. 12 In 1949, the combination of intestinal polyposis and pigmentation of the skin and mucous membranes was established as a distinct entity in a publication by Jeghers et al. 13 In 1954, Bruwer et al coined the eponym “Peutz–Jeghers syndrome” in the title of his article on this disorder. 14 The prevalence of PJS is 1 in 200,000. 15 PJS is characterized by mucocutaneous melanosis, gastrointestinal polyposis, intestinal, and extraintestinal cancers. 16 The disease may have variable penetrance within the same family. 17 Gastrointestinal PJS polyps are most common in the upper jejunum, followed by colon and stomach. 11 Gastric polyps occur in approximately 15%-30% of PJS cases. 4 Clinical manifestations include intussusception, chronic gastrointestinal bleeding, and anemia. The patients might be subjected to multiple laparotomies, putting them at risk for bowel obstruction. 17 On endoscopy, these polyps have a velvety or papillary surface and resemble hyperplastic polyps (Fig. 1A). 2 The polyps in colon often have a characteristic morphology with branching smooth muscles covered by hyperplastic epithelium giving rise to a characteristic arborizing (Christmas tree) pattern. Furthermore, lobulated clusters of colonic crypts, a feature that may discriminate them from other polyps such as hyperplastic polyps or juvenile polyps, can be seen. 18 However, gastric Peutz–Jeghers polyps often lack the typical arborizing histology pattern and are often not readily distinguished from gastric juvenile polyps or hyperplastic polyps (Fig. 1B). 19 Sometimes, displacement of mucinsecreting glands into the submucosa or muscularis propria can mimic well-differentiated adenocarcinoma. 20 Malignant transformation of the gastric PJS polyps, though rare, has been reported.21–23 In addition, Defago et al have described carcinoma in situ in a gastric polyp in a patient with PJS. 24 Though the overall cancer risk in PJS is highest for colorectal carcinoma, these patients have approximately 29% risk of developing stomach cancer. 25 The common extracolonic tumors in these patients include pancreas, breast, ovary (sex cord stromal tumors with annular tubules), testis (Sertoli cell tumors), and cervix (adenoma malignum). 20 Per the American College of Gastroenterology (ACG) guidelines, any individual with perioral/buccal pigmentation with at least two histologically confirmed PJS type polyps or family history should be tested for STK11 gene mutation for PJS. 26 The phenotype may be more severe in families with a truncating STK11 mutation than a missense mutation. 20 The recommended endoscopic surveillance interval for gastrointestinal manifestations is three years and for extraintestinal neoplasms is one year. 26

(
Solitary PJ polyps are rare. These polyps occur in the later stage of the life and are larger in size than the polyps in PJS. 27 The largest polyp described so far has been 15 cm in dimension. 27 The endoscopic and histologic appearances are similar to that of syndromic PJ polyps. 11 Some authors have suggested that the solitary gastric PJ polyps have less branching of the muscularis mucosae as compared with familial PJS polyps. 28 Jin et al also described a proliferation of smooth muscles in the submucosa. The cases of solitary PJ polyps were found negative for STK11 mutation, which is classically detected in PJS.29,30 Solitary PJ polyps have been known to have a good prognosis with no evidence of malignant transformation.22,31 Low-grade intraepithelial neoplasia has been rarely reported in cases of solitary PJ polyps.29,32 However, Burkart et al questioned the existence of true solitary PJ polyps and found a higher association of intestinal and extraintestinal malignancies in these patients. Due to this observation, they proposed that solitary PJ polyps may represent an early or incomplete form of PJS, and these patients may have similar lifetime risk of malignancy. 32
Juvenile Polyposis Syndrome Polyps
Juvenile polyposis syndrome (JPS) is the most common heritable gastrointestinal polyposis syndrome with a prevalence of 1 in 100,000-160,000. 33 It is an autosomal dominant disorder characterized by a mutation in SMAD4 (also called the MADH4 gene), BMPRA1, or ENG genes.11,19,26 All these genes are a part of the tumor growth factor-β signaling family and directly or indirectly affect the cell growth inhibition and apoptosis. The polyps most commonly affect the colon and rectum (98%), followed by stomach (14%), jejunum/ileum (7%), and duodenum (2%). 25 On endoscopic examination, the polyps have a rounded and smooth contour (Fig. 1C), in contrast to the papillary surface of PJ type polyps. On microscopic examination, 17 the polyps were found to be characterized by dilated mucous-filled glands in an edematous, inflamed lamina propria (Fig. 1D). These patients have 17%-22% risk of colorectal carcinoma and 10%-21% lifetime risk of gastric and duodenal carcinoma. Patients with SMAD4 mutations should also be screened for hereditary hemorrhagic telangiectasia, especially pulmonary arteriovenous malformation due to the risk of pulmonary hemorrhage.25,34 Lam-Himlin et al found in their study that the features of gastric JP type and PJ type polyps are less specific than their intestinal counterparts, and there may be a significant morphologic overlap between the two groups. 19 Studies have shown that SMAD4 mutation is more commonly associated with epithelial phenotype with high crypt density, while BMPRA1 is more frequently associated with stroma-rich phenotype. 35 While van Hattem et al found no difference in the association of dysplasia with these two mutations, 35 Handra-Luca et al demonstrated that SMAD4 mutations are associated with a more aggressive polyp phenotype. 36 Gastric polyposis is more commonly seen in SMAD4 mutation-related JP.34,37 Per the ACG guidelines, any individual with five or more JPs in the colorectum or any number of JPs in the other parts of the gastrointestinal tract should be evaluated for JPS by testing for SMAD4 and BMPR1A gene mutations. In patients testing positive for JPS, endoscopic follow-up is recommended every one to three years. 26 Once a disease-causing mutation is identified in a patient with JPS, other family members should also undergo mutation-specific testing to determine whether the disease is present or absent so that appropriate surveillance can be undertaken. 26
Solitary JPs are common, encountered in around 2% pediatric population. 35 They are common in the colorectum but are rarely reported in the stomach and small bowel as well.8,15 Endoscopically and histologically, they are similar to syndromic JPs. 17 These are benign polyps with no associated risk of malignancy. However, multiple JPs (>3) should raise a possibility of JPS as it has a 39% lifetime risk of malignancy and requires surveillance.8,35
PTHS Polyps
PTHS is a group of autosomal dominant inheritable disorders characterized by mutations in the PTEN gene. 25 These include Cowden syndrome (CS), Bannayan–Riley–Ruvalcaba syndrome (BRRS), Proteus syndrome, and Proteus-like syndrome.16,38 Mutations in other genes, such as SDH (succinate dehydrogenase B, C, and D), PIK3CA, and AKT1, as well as hypermethylation of KLLN have been identified in a subset of PTEN mutation-negative patients.25,39
Cowden syndrome
The diagnosis of CS is made based on the International Cowden Consortium criteria, which were modified by the National Comprehensive Cancer Network. 11 The incidence of CS in the general population is 1 in 200,000 and more than 90% patients present in the adult life by the late third decade. 38 Mucocutaneous hamartomas (trichilemmomas, acral keratosis, and papillomatous lesions) are pathognomonic features of CS. 38 Macrocephaly and Lhermitte–Duclos disease or dysplastic cerebellar gangliocytoma are two other features considered specific for CS and are included in the major criteria.40,41 These patients have an increased risk of breast, thyroid, and endometrial carcinoma, which are the other major criteria. 40 Patients with CS also have a predisposition to benign hamartomatous outgrowths such as lipomas, arteriovenous malformations, fibrocystic breast disease, benign thyroid nodules, multiple uterine leiomyomas (fibroids) and/or bicornuate uterus, and gastrointestinal polyps. 40 Diffuse esophageal glycogenic acanthosis is present in more than 80% of CS patients and may be diagnostic for CS in the presence of other benign gastrointestinal polyposis.17,42,43 Gastrointestinal polyps occur in up to 50% of patients with CS with a wide variety of endoscopic and histologic features, including adenomatous, inflammatory, hyperplastic, lymphoid, ganglioneuromatous, and leiomyomatous polyps (Fig. 1E and F).11,44 The majority of CS patients (>50%) have two or more different polyp histologies. 44 Though most studies describe polyps in CS being colonic, gastric polyps are present in almost all patients with CS and are usually numerous with a variable appearance.42,45 Depending on the major histologic component, they can be smooth contoured or have a hyperplastic/papillary configuration endoscopically. The polyps in the stomach are commonly misdiagnosed as hyperplastic hamartomatous polyps.42,45 Though dysplasia has not been reported in gastric polyps in CS, patients with CS and gastric cancer have been reported.46–48 The risk of colorectal carcinoma in CS is 7%-15%, and 1 in 100 patients with CS may develop gastric malignancy. 11
Individuals with multiple gastrointestinal hamartomas or ganglioneuromas should be evaluated for PTEN gene mutation. The recommended gastrointestinal surveillance for patients with PTEN gene mutation is colonoscopy and esophagogastroduodenoscopy examination beginning at the age of 15 years and repeated every two years or two to three years, 26 though different suggestions has been suggested. 49
Bannayan–Riley–Ruvalcaba syndrome
BRRS is another PTEN-related congenital autosomal dominant syndrome characterized by macrocephaly, penile freckling, slowed psychomotor development along with multiple cutaneous, and visceral hamartomas such as lipomas and hemangiomas. Diagnostic criteria for BRRS have not been set but are based heavily on the presence of the above cardinal clinical features along with PTEN mutation. Intestinal polyposis affects approximately 45% of the affected individuals, and the polyp characteristics are similar to CS.17,50 Owing to the similarity of germline mutations in the PTEN gene, it has been proposed that CS and BRRS may be the same syndrome along a broad spectrum. The various mutations or deletions in the different regions of the PTEN gene may confer the varying risk for developing BRRS versus CS. 17
Proteus syndrome and Proteus-like syndromes
Proteus syndrome is a complex, highly variable disorder characterized by disproportionate growth of skin, skeleton, and central nervous system tissue.16,38 The specific diagnostic criteria include connective tissue nevi, epidermal nevi, dysregulated adipose tissue, vascular malformation, and facial phenotype. 51 Proteus-like syndrome is undefined but refers to individuals with significant clinical features of Proteus syndrome who do not meet the diagnostic criteria for Proteus syndrome. 38 The gastrointestinal findings reported in Proteus syndrome include rectal polyps, colonic lipomatosis, and gastrointestinal hemangiomas. 51 The rectal polyps described by Lamireau et al had an inflammatory polyp morphology with foci of ossification. 52
Hereditary Mixed Polyposis Syndrome Polyps
Hereditary mixed polyposis syndrome (HMPS) is a rare gastrointestinal polyposis syndrome that was originally described in a large Ashkenazi Jewish family with multiple colorectal polyps and cancer.53,54 It has been mapped to a locus on chromosome 15q13.3-q14 in a number of families with HMPS, but the exact underlying mechanism still remains clear. Recently, a duplication of approximately 40 kb upstream of the gremlin 1 (GREM1) gene that encodes the secreted bone morphogenetic protein (BMP) antagonist at chromosome 15 was found to be associated with HMPS.55,56 Increased GREM1 expression is predicted to cause reduced BMP pathway activity, a mechanism that also underlies tumorigenesis in juvenile polyposis of the large bowel. 55 The mean age of polyp occurrence in one family was 28 years. Polyps in HMPS patients have various different morphologies, including atypical juvenile polyps, hyperplastic polyps, and adenomas. 57 These glands may show a strikingly serrated morphology mimicking serrated adenoma/polyp. 20 HMPS may be misdiagnosed as JPS or serrated polyposis syndrome and vice versa. Though HMPS is rare and the exact course of polyp progression is not known, they are thought to follow a hyperplastic to juvenile to Peutz–Jeghers, then adenomatous and finally carcinoma sequence. 57 This disease appears to affect only the colon and does not involve the stomach or small intestine; no other accompanying extraintestinal manifestation have been currently described. 17 Genetic testing for GREM1 mutation and expression might be considered in families with adenomatous and hamartomatous polyposis in which an etiology cannot be determined. Based on the current knowledge of this entity, management should probably be similar to that for familial adenomatous polyposis (FAP). 26 Of note, a most recent study demonstrated that the prevalence of GREM1 mutation among Lynch syndrome Ashkenazim is 0.7%, and one mutation carrier was found who fulfills the Amsterdam criteria for Lynch syndrome. 56 The relationship between GREM1 mutation and Lynch syndrome is still unclear.
Cronkhite–Canada Syndrome Polyps
Though not really hereditary, multiple gastrointestinal polyps have been reported in Cronkhite–Canada syndrome (CCS). This rare protein-losing gastroenterocolopathy syndrome also has other clinical features, including chronic diarrhea, malnutrition, onycholysis, alopecia, and skin hyperpigmentation. 58 The majority of patients (>80%) are diagnosed at the age of 50 years or older, and the mean age at presentation is 59-63.5 years.59,60 The extremely rare pediatric cases reported actually have features of infantile juvenile polyposis.61,62 CCS has a poor prognosis, and the five-year mortality rate can be up to 50% if it is untreated or if treatment is delayed or inadequate. Although there is no standard therapy, limited success has been reported with antibiotics, steroids, nutritional therapy, 5-aminosalicylate acid, histamine H2 receptor antagonists, antitumor necrosis factor-α agents, immunomodulators, and eradication of Helicobacter pylori. 60 The precise mechanism of CCS remains elusive, but no convincing familial predisposition has been identified and to date no germline mutations have been associated with this disorder. Recent studies favor an autoimmune process characterized by immune dysregulation,63,64 and therefore, steroids are considered the mainstay of medical treatment, although the recommended dose and duration of their use have varied widely in the literature. 60
CCS polyps constitute less than 0.1% of the gastric polyps and resemble JPS polyps or hyperplastic polyps histologically characterized by expanded edematous lamina propria containing a predominantly mononuclear inflammatory cell infiltrate and tortuous, dilated to cystic glands/foveolae or crypts (Fig. 1G).1,59 Interestingly, in contradistinction to other GI polyposis syndromes, the intervening endoscopically/macroscopically spared nonaffected mucosa in CCS is histologically affected and shows lamina propria edema and inflammatory cell infiltration, as well as gland/crypt dilation and distortion.59,65 Usually, the polyps in CCS are diffuse throughout the entire gastrointestinal tract with esophagus sparing and are nonneoplastic; however, adenomatous transformation or dysplasia has been reported.65,66 There have been reports that colonic carcinomas can arise in the CCS polyps of colon.66,67 Gastric adenocarcinomas have also been reported in patients with CC polyps.68–70 At present, there is still no widely accepted algorithm or specific criteria on the diagnosis of CCS. The presence of diffuse gastrointestinal polyposis, characteristic histology in both endoscopically abnormal and abnormal mucosa, as well as relevant supporting clinical findings, including typical ectodermal changes, are the current cornerstones of diagnosis. 59
Fundic Gland Polyps
FGPs, the most common polyp types (13%-77% of all gastric polyps), 2 have been included in the hamartomatous group by some authors. Sporadic fundic gland polys have been associated with chronic proton pump inhibitor use. 71 Endoscopically, the polyps have a smooth, glassy, and transparent appearance and are usually multiple and present in the fundic region of the stomach. 2 Histologically, these polyps are characterized by dilated glands lined by parietal cells or mucous cells (Fig. 1H). 2 Dysplasia in sporadic FGPs is uncommon, and malignant transformation is rare. Levy and Bhattacharya reported low-grade dysplasia in 0.3% of all their FGPs with no progression to high-grade dysplasia or adenocarcinoma on follow-up. 71 There are occasional reports of high-grade dysplasia in sporadic FGPs. 72 FGPs are present in >80% of patients with FAP. Bianchi et al have found dysplasia in 41% of the FGPs associated with FAP. 73 The frequency of dysplasia was higher (62%) in the study by Arnason et al. 74 The frequency of high-grade dysplasia was similar in the two groups (3%-4%). Despite higher frequency of dysplasia in FAP-associated FGPs, gastric adenocarcinoma arising in FGPs is rare in FAP. 75 FAP-associated gastric polyps can show a hybrid phenotype, including foveolar hyperplastic, pyloric gland, and intestinal types.74,76 In our experience, these hybrid polyps can also resemble PJ type polyps morphologically. Dysplasia in FGPs should prompt a clinician to examine the patient for possible FAP or its variants. The recommended follow-up for patients who test positive for adenomatous polyposis coli (APC) gene mutation is endoscopic gastrointestinal surveillance every one to two years. 26
Gastric Inverted Hamartomatous Polyps
GIHPs are a distinct entity characterized by submucosal growth of hypertrophic glands with cystic dilatation.10,77 They are distinct from the other types of hamartomatous polyps, which have an exophytic configuration contrary to the endophytic nature of these polyps. 78 On endoscopic examination, these are reported as solitary submucosal masses. 79 On endoscopy, extrusion of milky mucinous material from the surface of the lesion and calcifications from the biopsy site may provide a clue to diagnosis.79,80 On histology, there is cystic proliferation of glands, which may be accompanied by smooth muscle proliferation, and formation of ectopic duct-like structures has also been reported.81,82 In addition, fibroblastic and neural proliferation may also be seen with glandular elements. 79 Diagnosis of GIHP is difficult without pathologic examination and may mimic ectopic pancreas on endoscopy and endosonography. 82 Certain features have been suggested on endoscopic ultrasound imaging, such as hyperechoic lesions with hypoechoic spots, which might be suggestive of GIHPs. 79 En bloc removal is recommended in lesions >2 cm due to the associated malignant risk (up to 20% risk of malignancy).83,84 Though it is rare, Hirasaki et al have reported a case of GHIP associated with signet ring cell carcinoma. 85
Other Syndromes Associated Hamartomatous Polyps
Other less common hereditary hamartomatous polyposis syndromes include Gorlin syndrome, neurofibromatosis type 1 (NF-1), and multiple endocrine neoplasia type 2b (MEN 2B). 17 Gastric polyposis has also been reported in McCune–Albright syndrome (MAS), a rare genetic disorder caused by postzygotic-activating mutation of the GNAS gene. MAS is characterized by the classical triad of skin hyperpigmentation (café-au-lait spots), polyostotic fibrous dysplasia, and endocrine dysfunctions, notably precocious puberty, hyperthyroidism, growth hormone excess, hyperprolactinemia, and hypercortisolism. The reported gastric polyp histology is similar to that of PJS. 86 Recently, the study on somatic GNAS mutation in GI tumors of PJS patients revealed that GNAS is not involved in the pathogenesis of GI tumors in PJS. 87 However, it is not clear whether gastric polyps are indeed a specific manifestation of these syndromes, though one would assume it might be possible. Further studies and more cases are needed to establish these relationships.
Conclusion
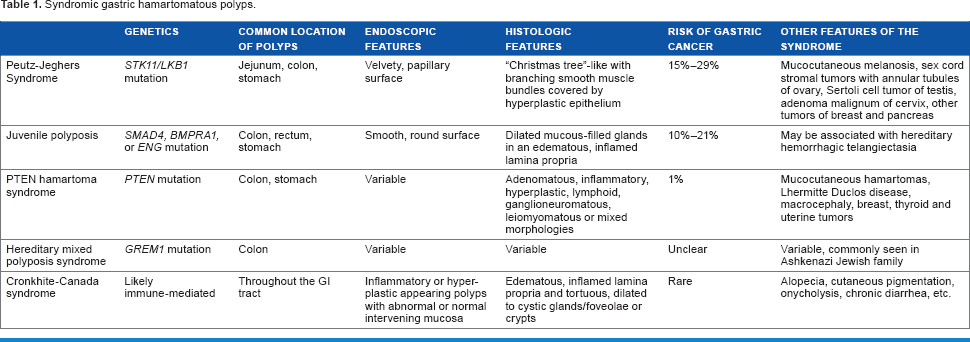
Hamartomatous polyps in the stomach are rare entities with variable clinical, endoscopic, and histologic features, genetic alterations, and risks of malignancy (Table 1). Multiple hamartomatous polyps should alert the pathologist and the clinician of the possibility of a hereditary polyp/cancer syndrome. There is an overlap in the morphologic features of various hamartomatous polyps, and molecular genetic testing is essential to establish the definitive diagnosis in the proper clinical context. Though the risk of malignant transformation is rare in hamartomatous polyps, some polyps including GIHPs and polyps in syndromes including PJS and JPS have a higher risk of malignant transformation. Collaboration between the pathologist, clinician, and genetic counselor is essential in making a diagnosis of polyposis syndromes. This is beneficial not only to patients for initiation of early surveillance but also for testing of family members for polyposis syndromes.
Syndromic gastric hamartomatous polyps.
Author Contributions
Conceived the concepts: MV and XZ. Analyzed the data: MV. Wrote the first draft of the manuscript: MV. Contributed to the writing of the manuscript: MV, XY and XZ. Agree with manuscript results and conclusions: MV, XY and XZ. Jointly developed the structure and arguments for the paper: XY. Made critical revisions and approved final version: MV, XY and XZ. All authors reviewed and approved of the final manuscript.