
Abstract
Cyclooxygenase (COX) plays a critical role in peptic ulcer development. COX-2 contains CpG islands in promoter area, which suggests possible epigenetic mechanisms of gene silencing. We evaluated COX-2 gene promoter methylation levels in the gastric mucosa of patients with various gastric diseases. DNA was extracted from endoscopic biopsy materials collected from the gastric mucosa. The methylation levels of the COX-2 gene promoter were measured quantitatively by using pyrosequencing. COX-2 mRNA expression in Kato III and AGS cells was measured using real-time PCR. COX-2 gene promoter methylation levels were significantly higher in Helicobacter pylori (HP)-positive cases than in HP-negative cases (27.5% vs. 8.1%, respectively, P < 0.001). COX-2 gene promoter methylation levels in patients in whom HP was successfully eradicated were significantly lower than those in HP-positive cases (18.7% vs. 27.5%, respectively, P < 0.01). We then investigated the effects of COX-2 gene promoter methylation on its mRNA expression in vitro. COX-2 mRNA expression was not observed in Kato III cells, despite the addition of the protein kinase C stimulator a-phorbol 12,13-dibutyrate (PDBu). COX-2 expression was observed after the addition of the demethylating agent 5-Aza-dC and was enhanced by PDBu. HP infection caused a significant increase in the methylation levels of the COX-2 gene promoter in the gastric mucosa. In addition to transcriptional regulation, COX-2 expression is regulated through epigenetic mechanisms.
Keywords
Introduction
Cyclooxygenase (COX) plays a pivotal role in the gastric mucosal barrier system. 1 3 Two major isoforms of COX have been identified. In the stomach, prostanoids synthesized via the constitutively expressed COX-1 pathway are responsible for cytoprotection of the gastric mucosa, whereas COX-2 mRNA levels increase rapidly in response to inflammatory and mitogenic stimuli. Non-selective COX inhibitors damage the gastrointestinal (GI) mucosa, and GI injury represents the most significant clinical side effect of chronic non-steroidal anti-inflammatory drug (NSAID) use. 4 Thus, selective COX-2 inhibitors were expected to be efficacious anti-inflammatory drugs without the side effect of GI toxicity. However, clinical trials suggest that selective COX-2 inhibitors still result in severe GI ulcer complications, albeit at approximately half the rate of conventional NSAIDs.5,6 In support of this, many recent animal studies have suggested that in contrast to the initial concept, COX-2 contributes to the repair of gastric mucosal damage.7,8 Thus, COX-1 and COX-2 play pivotal roles in gastric mucosal defense mechanisms.
The aberrant methylation of 5′-CpG islands (CGIs) has been implicated in transcriptional silencing of various genes involved in aging and cancer. 9 The 5′-region of COX-2 has a typical CGI; accordingly, COX-2 protein expression is strongly suppressed by gene promoter methylation in several gastric neoplasms. Helicobacter pylori (HP) infection causes aberrant DNA methylation in various genes, including COX-2, in the gastric mucosa. 10 13 The expression of COX-2 in the injured gastric mucosa plays a significant role in the repair process. In the present study, we evaluated COX-2 gene promoter methylation levels in the gastric mucosa of patients with various gastric diseases using quantitative bisulfite-pyrosequencing. We also investigated the transcriptional regulation of COX-2 expression through DNA methylation in vitro. Our findings indicate that COX-2 is densely methylated in HP-infected gastric mucosa and there may be a relationship between COX-2 gene promoter methylation and gastric mucosal damage.
Methods
Sample Collection
Endoscopic examinations were performed at St. Marianna University School of Medicine Hospital (Kanagawa, Japan) from March 2009 to December 2011. DNA was extracted from the endoscopic biopsy samples with informed consent. In HP gastritis, gastric atrophy typically begins in the antrum and progresses toward the gastric body. In the gastric body, HP-associated mucosal inflammation is usually higher in the greater curvature than in the lesser curvature. To observe the maximum effect of HP-associated mucosal inflammation on COX-2 gene promoter methylation, we collected specimens from the gastric antrum and body of the greater curvature. The samples were centrifuged immediately and the pellets were frozen at -80 °C. DNA was extracted using the standard phenol-chloroform method. HP infection status was confirmed using a rapid urease test kit (Pylori Tek Test Kit; Eidia, Tokyo, Japan). The study protocol was approved by the institutional review board of St. Marianna University School of Medicine.
Bisulfite PCR and pyrosequencing analysis of DNA methylation
PCR and sequencing primers.

Cell culture
Kato-III cells, a poorly differentiated human gastric adenocarcinoma cell line, were maintained in RPMI-1640 medium. AGS cells, a well-differentiated human gastric carcinoma cell line, were maintained in Dulbecco's modified Eagle's medium. All cultures were supplemented with 10% fetal calf serum, 20 mM HEPES, 10 mM NaHCO3, and antibiotics, and were incubated at 37 °C under 95% air and 5% CO2.
Reverse-transcription PCR
COX-2 mRNA expression was measured by real-time PCR in cell lines with or without 5-Aza-dC or trichostatin A (TSA). First-strand cDNA was prepared by reverse transcription of 5 μg total RNA using Superscript III reverse transcriptase (Applied Biosystems, Foster City, CA, USA). Real-time quantitative reverse transcription PCR was carried out using TaqMan Gene Expression Assays (COX-2, Hs01573472 g1; glyceral-dehyde-3-phosphate dehydrogenase, Hs00266705 gl; Applied Biosystems) with a 7500 Real-time PCR System (Applied Biosystems) according to the manufacturer's instructions. SDS2.1 software (Applied Biosystems) was used to perform comparative ΔCt analysis. Glyceraldehyde-3-phosphate dehydrogenase served as an endogenous control.
Results
COX-2 gene promoter methylation levels in HP-positive patients
The clinical features of the controls and patients with gastritis, peptic ulcer, and a history of successful HP eradication are listed in Table 2. COX-2 has CG-rich CGIs in its promoter region. Previously, we reported that the methylation levels of several cancer-related genes in biopsy specimens and gastric wash samples from patients with gastric cancer showed a close-dependent relationship.
14
However, after preliminary experiment, we could not detect significant COX-2 gene promoter methylation in the gastric wash samples, even in patients for whom biopsy specimens showed dense COX-2 gene promoter methylation (Fig. 1A). Although cancer cells from the mucosal layer appear to be easily exfoliated into gastric washes, the exfoliation of normal mucosal cells may be limited and the DNA recovered from our gastric washes was relatively degraded. Therefore, we used endoscopic biopsy samples for further analyses. We then compared the COX-2 gene promoter methylation levels in samples from different locations in the stomach. In patients positive for COX-2 gene promoter methylation (levels >15%), no significant difference was observed between samples from the greater curvature of the gastric middle body and the greater curvature of the antrum (Fig. 1B). Therefore, samples from the greater curvature of the gastric antrum were used in subsequent experiments.
( Clinical features of patients.
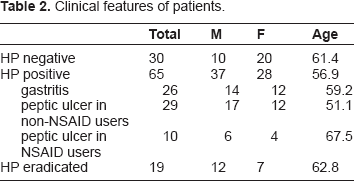
First, we compared COX-2 gene promoter methylation levels in the gastric mucosa between HP-positive and HP-negative cases (n = 95). HP-eradicated cases or patients with a gastric neoplasm were not included in this experiment. As reported previously in qualitative experiments,
14
COX-2 gene promoter methylation levels were significantly higher in HP-positive cases than in HP-negative cases (27.5% vs. 8.1%, respectively, P < 0.001; Fig. 2). The average ages of the HP-positive and HP-negative groups were not significantly different (63.7 years vs. 61.3 years, respectively, P = 0.578), indicating that the differences observed were not due to age-related methylation. HP-positive cases included HP gastritis (n = 26) and active peptic ulcer (n = 39), and we observed dense COX-2 gene promoter methylation in both groups (26.7% and 28.4%, respectively) with no significant difference between groups (P = 0.26). Among HP-positive peptic ulcer patients, 10 patients were NSAID users. COX-2 gene promoter methylation levels in the patients with HP-positive peptic ulcers were 29.6% in non-NSAIDs users compared to 27.9% in NSAID users (P = 0.45).
COX-2 gene promoter methylation levels in patients with or without HP infection, or in patients after HP eradication. The values are the mean ± SE. Statistical analysis was performed using Student's t-test.
Next, we examined the effect of HP eradication on COX-2 gene promoter methylation. We examined patients who were previously successfully treated (>1 year prior to examination; n = 19) for HP infection. COX-2 gene promoter methylation levels in these patients were significantly lower than in the HP-positive cases (18.7% vs. 27.5%, respectively, P < 0.001; Fig. 2). These results indicate that HP infection causes reversible COX-2 gene promoter methylation in the gastric mucosa.
Effects of COX-2 gene promoter methylation on its mRNA expression in vitro
We then examined the effects of COX-2 gene promoter methylation on its mRNA expression in vitro using the human gastric carcinoma cell line Kato III. In these cells, the COX-2 gene promoter is densely methylated
10
and they do not express COX-2 mRNA.
15
We also used a well-differentiated human gastric adenocarcinoma cell line AGS in which COX-2 gene promoter is moderately methylated.
16
COX-2 is an immediate-early gene induced by cytokines, growth factors, and tumor promoters. Reports indicate that phorbol ester induces COX-2 mRNA expression in several gastric cancer cell lines;
17
however, we did not observe expression in Kato III cells, even after the addition of the protein kinase C stimulator α-phorbol 12,13-dibutyrate (PDBu) (Fig. 3A).
Effects of a PKC stimulator (α-phorbol 12,13-dibutyrate; PDBu) on COX-2 mRNA expression with or without 5-aza-dC in KATO-III cells (
To examine the role of methylation and histone deacetylation in the silencing of COX-2, we treated Kato III cells with 5-aza-dC, a methyltransferase inhibitor, or TSA, a histone deacetylase inhibitor. Treatment with 5-aza-dC effectively decreased methylation levels from 89% to 65% in Kato III cells. Notably, COX-2 mRNA expression levels were restored after the addition of 5-aza-dC and were further enhanced by treatment with PDBu. In contrast, treatment with TSA did not induce COX-2 mRNA expression, even in the presence of PDBu (data not shown). In AGS cells, treatment with 5-aza-dC decreased methylation levels from 45% to 40%. A modest increase in COX-2 mRNA expression was observed after 5-aza-dC treatment in AGS cells (Fig. 3B). These results indicate that COX-2 mRNA expression is regulated through transcriptional and DNA methylation mechanisms. Histone acetylation did not appear to be involved in silencing COX-2 expression in these cells.
Discussion
Using quantitative pyrosequencing, we demonstrated that COX-2 gene promoter methylation levels in the gastric antral mucosa of HP-positive patients were significantly higher than in HP-negative cases. Moreover, COX-2 gene promoter methylation levels in HP-eradicated cases were significantly lower than in HP-infected cases. In vitro experiments using KATO III and AGS cells showed that, in addition to transcriptional regulation, COX-2 expression was regulated through an epigenetic mechanism.
COX is a membrane-bound glycoprotein that functions as a rate-limiting enzyme in prostaglandin synthesis. COX-1 and COX-2 are the 2 major COX enzyme isoforms. 1 3 In human gastric mucosa, COX-1 and COX-2 immunoreactivity is localized mainly in parietal cells, with some in macrophages and myofibroblasts. 18 Previously, it was thought that only COX-1 participated in gastric mucosal defense; however, several clinical trials have suggested that a COX-2 selective inhibitor produces less gastrointestinal toxicity compared with traditional NSAIDs.5,6 In agreement with this, animal studies have suggested that COX-1 and COX-2 are both necessary for gastric mucosal healing. COX-1 inhibition alone, which can be induced pharmacologically via specific inhibitors or genetically via gene targeting, 19 does not cause gastric mucosal injury. A combination of selective inhibitors for COX-1 and COX-2 is required to cause hemorrhagic erosion in the gastric mucosa comparable to that seen with indomethacin. COX-2 mRNA and protein expression increases during repair of gastric mucosal lesions, and selective COX-2 inhibitors delay mucosal healing in rats and mice.7,20 The contribution of COX-2 to total prostaglandin synthesis in the stomach appears to be very small; however, it is very important in terms of mucosal defense. 8 A variety of growth factors (HGF, EGF, TGFa, and VEGF) appear to be induced in diverse COX-2-dependent ulcer repair processes. 3 Overall, the inhibition of COX-2 activity appears to interfere with ulcer healing.
Epigenetics refers to a variety of processes that have long-term effects on gene expression without changing the DNA sequence. 9 The key players in epigenetic control are DNA methylation and histone modifications. DNA methylation is associated with gene silencing and occurs on cytosines at CpG dinucleotides across the human genome. However, although it has been hypothesized that DNA methylation is fixed for a generation, it seems that not all DNA methylation remains stable and that patterns result from a balance of methylating and demethylating activity. 21 Chronic inflammation, including HP infection, causes aberrant DNA methylation. 22 HP infection in humans is best modeled in Mongolian gerbils. HP infection induces aberrant DNA methylation of several CGIs in Mongolian gerbils. 23 The immunosuppressive drug cyclosporine A blocks inflammation of the gastric mucosa and induction of altered DNA methylation in this model organism. Thus, DNA methylation alterations that occur in the gastric mucosa after HP infection result from an infection-associated inflammatory response, and are not directly due to HP.
Previous qualitative studies have revealed aberrant methylation of COX-2 in HP-infected gastric mucosa and in gastric and colorectal cancers.10,13 Using a highly sensitive quantitative method, we observed that COX-2 DNA from non-cancerous HP-positive gastric mucosa in patients with gastritis, peptic ulcers, and NSAID-induced peptic ulcers was densely methylated. Immunohistochemical studies showed that COX-2 localizes to human epithelial cells in HP-infected gastric mucosa. 17 Our in vitro experiments suggest that COX-2 gene promoter methylation may result in reduced COX-2 mRNA induction. Taken together, COX-2 gene promoter methylation may be involved in the vulnerability of HP-infected patients to gastric mucosal damage. Several clinical studies have found that HP infection and NSAID use independently increase the risk of peptic-ulcer disease and ulcer bleeding. 24 26 When HP-infected patients use an NSAID or aspirin, which inhibit COX-1 activity, the actions of both COX-1 and COX-2 may be impaired, thus enhancing the risk of developing mucosal damage. In this study, we also demonstrated that COX-2 gene promoter methylation levels in patients in whom HP has been eradicated were significantly lower than those of HP-positive cases. Since COX-2 is associated with mucosal healing, these results support clinical findings that NSAID-induced ulcer risk and HP-associated ulcer recurrence rates are reduced after HP eradication. 26 28
In summary, the COX-2 gene promoter is densely methylated in the gastric mucosa of HP-infected patients. COX-2 mRNA expression is under transcriptional and epigenetic control. COX-2 gene promoter methylation could be involved in ulcerogenesis in HP-infected patients.
Author Contributions
Conceived and designed the experiments: YM, HY, YW. Analyzed the data: YM, HY, YW. Wrote the first draft of the manuscript: YM, HY, YW. Contributed to the writing of the manuscript: YM, HY, YW, RO, YO, TM, FI. Agree with manuscript results and conclusions: YM, HY, YW, RO, YO, TM, FI. Jointly developed the structure and arguments for the paper: FI. Made critical revisions and approved final version: YM, HY. All authors reviewed and approved of the final manuscript.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, and Culture of Japan to HY.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Footnotes
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.