
Abstract
The efficacy of approaches in which vascular disrupting agents (VDA) are used in combination with conventional chemotherapy and/or radiation therapy in the treatment of cancer might be improved if there were a better understanding of the cellular and molecular changes induced in normal and malignant cells as a result of VD A exposure. Toward this goal, murine endothelial cells were treated
Introduction
Vascular disrupting agents (VDAs) provide a promising new approach to the treatment of cancer. Unlike conventional chemotherapeutics, most of which act directly on tumour cells, VDAs instead target the blood vessels upon which the survival and growth of a tumour mass depends. 1 Of the VDAs that have so far progressed to human trials, most, including ZD6126, combretastatin A4 (CA4P), MN029 and AVE8062E, bind to and disrupt tubulin.1–3 They mediate their effects by inducing the immature endothelial cells lining the structurally abnormal blood vessels that supply a growing tumour mass to round up and detach from the basement membrane. 4 Intravascular coagulation is induced leading to vessel blockage and the cessation of nutritive blood flow. 4 Without the oxygen and nutrients required to maintain viability, a massive necrotic response quickly results particularly within central regions of a tumour mass. 4 Specificity is conferred by the fact that while endothelial cells in immature blood vessels rely on a tubulin cytoskeleton for the maintenance of their elongated shape, in more mature non-proliferating endothelial cells this function is largely supplanted by actin.5–7 Importantly, in contrast to established cytotoxic agents such as vinblastine or colchicine that bind to and destabilize tubulin or microtubule stabilizing cytotoxins such as paclitaxel and docetaxel, the depolymerizing activity of VDAs is rapidly reversible.4,8 As the compounds also have a relatively short plasma elimination half-life following intravenous administration,9–11 the toxicities commonly associated with the use of tubulin-directed anti-mitotic drugs are not seen with VDAs.11,12
A characteristic of VDA therapy, however, is the persistence around the edge of a treated tumour of a thin layer of viable cells that are believed to survive because they obtain their oxygen and nutrients, not from the tumour-associated neovasculature, but instead by diffusion from unaffected normal vessels present in surrounding non-malignant tissues.8,13 In the absence of further treatment, this so called “viable rim” can serve as a reservoir from which malignant cells can invade and repopulate the necrotic central regions of a treated tumour.8,13 Thus, VDAs are generally most effective when used in combination with conventional cytotoxic agents or radiation therapy that kill the comparatively well-oxygenated and mitotically active cells remaining within the viable rim.14–16
Up to this point the nature and timing of the regimens in which VDAs are used together with conventional cancer therapies have been determined largely empirically, guided mostly by physiologic considerations.17–24 Often overlooked is the fact that exposure of cells in the viable rim to a VDA might alter, in a positive or negative manner, their sensitivity to subsequent treatment with ionizing radiation and/or chemotherapeutic agents. If so, then understanding the cellular and molecular changes induced in both normal and malignant cells by VDA treatment may prove key to the design of more effective combination therapies involving VDAs and other modalities.
Our initial efforts in this regard have focused on the impact of VDA treatment on the radiation sensitivity of the endothelial cell component of the viable rim. The survival and proliferation of tumour cells that repopulate areas of necrosis following VDA treatment is critically dependent upon the re-establishment of an effective vascular supply and treatments that prevent angiogenesis have been shown to improve the efficacy of VDA treatment in initial studies.25,26 The spatial and temporal nature of radiation therapy makes it particularly attractive in this regard.
In the present study, we demonstrate that while brief exposure to ANG501, a novel stilbene developed in our laboratory that is closely related to the tubulin depolymerizing VDA combretastatin-A4, had little or no effect on the long-term survival and proliferation of murine endothelial cells (SVEC)
Materials and Methods
The ANG500 series
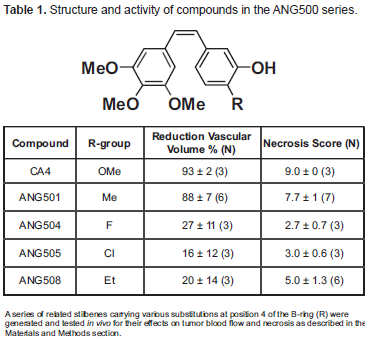
The ANG500 series includes a number of novel stilbenes closely related to the well-characterized VDA combretastatin-A4 differing from it only as a result of the replacement of the methyoxy group at position 4 of the B-ring with a variety of other groups (Table 1).
Structure and activity of compounds in the ANG500 series.
Vascular disrupting and anti-tumour activity of ANG501 and related compounds in vivo
The
Briefly, tumours were initiated by the subcutaneous injection of 50 μl of a crude tumour cell suspension into the flank of 12–16 week old syngeneic CBA/Gy fTO mice. Animals were selected for treatment when their tumours achieved a mean geometric diameter of approximately 5–6 mm (150–300 mg).
The effect of drug treatment on perfused/functional vascular volume was assessed using the fluorescent perivascular stain Hoechst 33342. Although VDAs are characterized by rapidly reversible tubulin depolymerizing activity, the vascular occlusion that results is longer lasting and slow to resolve. While maximal reduction in blood flow is generally evident 4–6 hr after VDA treatment, the degree of recovery by 24 hr is fairly modest. 30 Studies in which ischemia was induced by applying metal clamps across the base of subcutaneously implanted tumours indicated that greater than 99% of tumour cells were killed if blood flow was interrupted for 2 hr.31,32 Although, early studies suggested that at least 15 hr of vessel occlusion was necessary to achieve long-term tumour control, 33 in the case of C3H mammary tumours maintained at 37 °C, a 6 hr period of ischemia was curative in 3 of 7 treated tumours.31,32 Similar results have been reported for the CaNT tumour used in the present study. 34 Based on these considerations, a 6 hr time point was chosen to evaluate the vascular disrupting activity of the various compounds in the ANG500 series, while tumour necrosis was determined at 24 hr.
Briefly, 100 mg/kg of each compound were injected i.v. into groups of tumour-bearing animals. Although this particular dose is less than 1/5 MTD for both CA4P 8 and ANG510 (data not shown), little additional benefit was seen when higher concentrations drug were used. 8 Six hr after VDA administration, the mice received an i.v. injection of Hoechst 33342. They were sacrificed approximately 1 min later, their tumours excised and frozen sections prepared and examined. Percent vascular volume was determined by examination under UV irradiation using a Chalkley grid and point scoring system as previously described. 29
For VDAs to be effective, it is necessary that the vascular shutdown they produce is of sufficient magnitude and duration to trigger substantial tumour cell death. To determine whether this is the case with respect to the various members of the ANG500 series, tumours were excised from animals treated 24 hr earlier by i.v. injection of 100 mg/kg of each drug and fixed in 10% buffered formalin. After standard paraffin wax processing, 5 μm sections were cut, stained with hematoxylin and eosin and scored subjectively for necrosis in blinded fashion as previously described, 13 using the following scale: 0%–10% necrosis = Grade 1; 11%–20% = Grade 2; 21%–30% = Grade 3; 31%–40% = Grade 4; 41%–50% = Grade 5; 51%–60% = Grade 6; 61%–70% = Grade 7; 71%–80% = Grade 8; 81%–90% = Grade 9; and 91%–100% = Grade 10.
Effect of ANG501 on endothelial cell survival and proliferation
The SV40 (strain 4A) transformed murine axillary lymph node-derived endothelial cell line SVEC4-10 was purchased from the American Type Culture Collection (ATCC) (Manassas, VA). Cells were maintained at 37 °C in an atmosphere containing 5% CO2 in Dulbecco's Modified Eagles Medium supplemented with 10% bovine serum, 2 mM L-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin sulfate (DMEM + 10% FBS).
A standard clonogenic assay was used to determine the effect of ANG501 treatment on the long-term survival and proliferative potential of SVEC cells. Briefly, serial dilutions of ANG501 were added to log phase SVEC cultures and the plates incubated at 37 °C for 30 min. Adherent and non-adherent cells were then harvested and pooled. Treated cells were washed 3 times in Hanks balanced salt solution (HBSS) and resuspended in DMEM + 10% FCS. After careful counting, aliquots of each cell suspension were added to 10 cm tissue culture plates and medium added to a final volume of 10 ml. Plates were incubated undisturbed at 37 °C in an atmosphere containing 5% CO2. Approximately 14 days later, plates were fixed and stained with a solution containing 1% methylene blue in methanol and the number of colonies determined using a ColCount Imager (Oxford Optronix Ltd., Oxford, U.K).
Microarray analysis
Genome wide microarray analysis was carried out to identify genes induced by treatment with ANG501. Briefly, RNAs isolated from control SVEC and SVEC treated 4 hr previously with ANG501 (50 μg/ml, 30 min) were compared using a two color (Cy3 and Cy5) approach and Agilent Whole Mouse Genome Oligo Microarrays (SA Biosciencies, Frederick, MD). More than 41,000 genes and transcripts encompassing the entire mouse genome were represented on the arrays with enriched content sourced from UCSC, RefSeq, RIKEN, NIA, Ensemb, UCSC Goldenpath and Unigene databases. Signal data was collected using an Agilent 2565A Fluorescent Scanner (SA Biosciencies, Frederick, MD).
Western blot analysis
Polyclonal rabbit antibodies directed against heat shock protein 25 (HSP 25), HSP 40 and alpha B-crystallin (Cryab) and mouse monoclonal antibodies (mAbs) directed against HSP70 and p21Wafl were purchased from Stressgen/Assay Designs (Ann Arbor, MI).
Alexa Fluor® 680-conjugated goat anti-mouse IgG (H + L) and affinity-purified IRDye800-conjugated affinity-purified donkey anti-rabbit IgG secondary antibodies were purchased respectively from Invitrogen (Carlsbad, CA) and Rockland Immunochemicals, Inc., (Gilbertsville, PA).
Briefly, SVEC cells were harvested at various time points after treatment with ANG501 (50 μg/ml for 30 min), washed in PBS and solubilized by incubating the cell pellets for 15 min on ice at approximately 2 × 107 cells/ml in a lysis buffer containing 10 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% NP-40. Nuclei and detergent insoluble material were removed by centrifugation and the lysates stored at –80 °C until required. Thawed samples were added to an equal volume of non-reducing sample buffer (125 mM Tris, 20% (v/v) glycerol, 4.6% (w/v) SDS, pH 6.8) and incubated at 100 °C for 5 min. Total cellular proteins were separated by SDS-PAGE and transferred onto nitrocellulose membranes. The filters were incubated overnight at 4 °C in PBS containing 5% (w/v) milk protein then incubated for 8–12 hr with monoclonal or polyclonal antibodies directed against proteins of interest. After extensive washing, filters were incubated for a further 1–2 hr with an appropriate fluorescent-labeled secondary antibody. After additional washing, protein products were visualized using a Odyssey Infrared Imager (LI-COR Biotechnology, Lincoln, NE).
Cell cycle analysis
SVEC cells were harvested by trypsinization at various time points after treatment with ANG501 (50 μg/ml for 30 min). The cells were washed twice in PBS and the pellets resuspended at approximately 1 × 106 cells/ml in 70% ethanol. Samples were stored at –20 °C until all time points had been collected. Fixed cells were pelleted by centrifugation at 1000 rpm for 10 min then resuspended with gentle mixing at approximately 1 × 106/ml in a staining solution containing 500 μg/ml propridium iodide and 100 μg/ml RNase A. Cells were stained for a minimum of 4 hr then analyzed using a FACScan (BD Biosciences, Immunocytometry Systems, Mountain View, CA). Cell cycle phase distributions were determined using the CellFit software package.
Effect of ANG501 treatment on the radiosensitivity of endothelial cells
SVEC cells treated 6 hr earlier with ANG501 (50 μg/ml, 30 min) to induce cell cycle arrest were exposed to a range of radiation doses between 1–10 Gy and the percentage of surviving cells determined using a clonogenic assay as described above.
Results
Vascular disrupting ability of ANG501
Previous structure/function studies have suggested that the
Effect of ANG501 on endothelial cell morphology and adherence
ANG501 induces rapid changes in endothelial cell morphology. As shown in Figure 1, even brief exposure to the drug caused SVEC to round up and detach from the plastic surface upon which they were growing. Changes were evident as early as 5 min after the addition of ANG501 and virtually all cells were non-adherent after 1 hr of treatment. Importantly, the effect was reversible and upon removal of ANG501, treated cells rapidly reattached and quickly regained their normal extended morphology (Fig. 1).
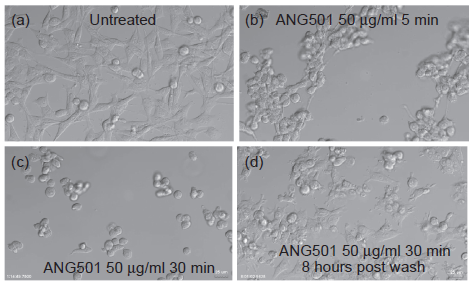
Effect of ANG501 treatment on endothelial cell morphology and adherence. ANG501 was added to non-confluent monolayers of SVEC cells at a final concentration of 50 μg/ml and digital images collected regular intervals. After 30 min incubation at 37 °C, cells were harvested by gentle pipetting, washed to remove drug, replated and additional images were collected.
Anti-mitotic activity of ANG501
Although treatment with ANG501 induces dramatic changes in the shape and adherence properties of endothelial cells (Fig. 1), exposure to the drug at concentrations up to 100 μg/ml for 30 min, had little or no effect on long-term viability, as determined using a standard clonogenic assay (Fig. 2a). Mean colony area was also not reduced, suggesting that the proliferative potential of individual clonogens was not significantly impacted by drug treatment (Fig. 2b).
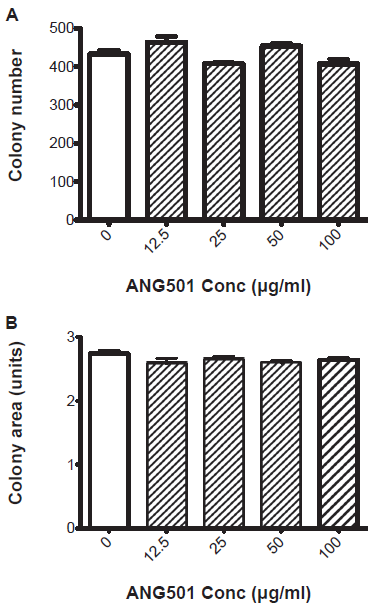
Cytotoxic activity of ANG501. A standard clonogenic assay was used to determine the effect of ANG501 treatment on the survival and proliferation of endothelial cells. Briefly, serial dilutions of ANG501 were added to log phase SVEC cultures and the plates incubated at 37 °C for 30 min. Adherent and non-adherent cells were then harvested and pooled and aliquots of the resultant cell suspension plated in 10 cm tissue culture dishes. Cultures were incubated undisturbed for approximately 14 days later after which they were fixed and stained with a solution containing 1% methylene blue in methanol. The total number (
Effect of ANG501 on endothelial cell gene expression
In order to identify changes induced by ANG501 treatment that could potentially modify the response of endothelial cells to ionizing radiation and/or other modalities, genome wide microarray analysis was carried out comparing RNA isolated from control cells with RNA isolated from SVEC 4 hr after treatment with ANG501 (50 μg/ml, 30 min). Among the functionally important genes that appeared to show elevated expression relative to untreated cells were the heat shock proteins HSP25, HSP70 and anti-B crystallin and the cell cycle regulator p21Wafl (data not shown). Western blot analysis confirmed that the changes observed at the RNA level were reflected by corresponding alterations in protein expression (Fig. 3). Of particular interest, p21Wafl which is barely detectable in untreated SVEC is rapidly induced following exposure to ANG501, with maximum expression occurring around 4 hr and returning to near pre-treatment levels by 24 hr (Fig. 3).
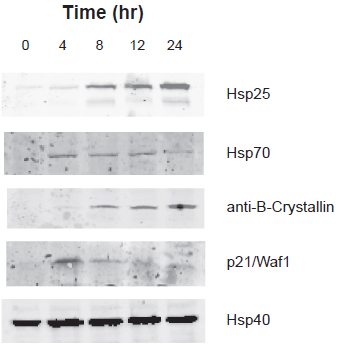
Effect of ANG501 on endothelial cell gene expression. Alterations in the expression of genes of interest upon ANG501 treatment (50 μg/ml for 30 min) was determined by Western blot analysis.
Effect of ANG501 on cell cycle
p21Wafl is a key regulator of the cell cycle. Correlating with the induction of p21Waf-1, brief exposure to ANG501 (50 μg/ml, 30 min) caused proliferating endothelial cells to accumulate in G2/M. The effect was transient. Although almost all cells were blocked at G2/M 6 hr after treatment (Fig. 4), by 24 hr normal cycling had largely resumed (Fig. 4).
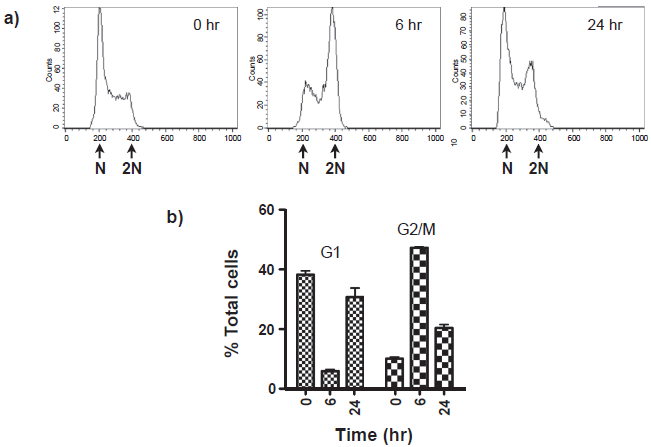
Effect of ANG501 on endothelial cell cycle progression. SVEC cells harvested at various time points after treatment with ANG501 (50 μg/ml for 30 min) were fixed in ethanol and stained with a solution containing 500 μg/ml propridium iodide and 100 μg/ml RNase A. DNA content a) was determined by FACScan analysis. Cell cycle phase distributions b) were estimated using the CellFit software package.
Effect of ANG501 on radiosensitivity
In general, cells in G2/M show elevated sensitivity to ionizing radiation. Reflecting this fact, clonogenic assays demonstrated that the survival of endothelial cells following exposure to radiation was reduced if they were treated 6 hr earlier with ANG501 (50 μg/ml, 30 min) (Fig. 5).
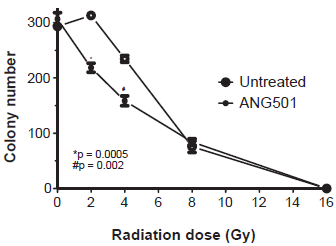
Effect of ANG501 on radiosensitivity. SVEC cells treated 6 hr earlier with ANG501 (50 μg/ml for 30 min) to induce cell cycle arrest were exposed to a range of radiation doses between 1–10 Gy and a clonogenic assay used to determine the impact of treatment on cell survival and proliferation.
Importantly, the radiosensitizing effect of ANG501 was statistically significant at clinically relevant radiation doses (p = 0.0005 and p = 0.002 at 2 and 4 Gy respectively) (Fig. 5).
Discussion
Although VDAs induce substantial intratumoural necrosis, their effect on tumour growth when used in a monotherapy setting is often rather modest. This apparent paradox can be explained by the presence around the edge of a treated tumour of a thin layer of viable cells that are believed to survive because they receive the oxygen and nutrients they require to remain viable from established vessels in surrounding normal tissues, which unlike the structurally immature vessels found within tumours, are not affected by VDA treatment.8,13 Unless prevented by additional treatment, the cells present in this “viable rim” can proliferate and rapidly repopulate necrotic regions of a treated tumour allowing progressive growth to resume.8,13 It is for this reason that VDAs are generally most effective when used in combination with other therapies. Thus far, most of the approaches that have been used in conjunction with VDAs were chosen because of their assumed activity against the malignant cell population. It was reasoned that since tumour cells in the viable rim are relatively well-oxygenated and have a high mitotic index they will be relatively sensitive to conventional cytotoxic therapies.35–40 However, since successful repopulation of the necrotic areas of a VDA-treated tumour requires the provision of adequate levels of oxygen and nutrients, it follows that this process will be critically dependent upon revascularization. Clearly, if angiogenesis can be inhibited, then the rapid regrowth of tumours that often occurs after VDA treatment might be slowed and/or prevented. In support of this conclusion, animal studies in which VDA treatment was followed by anti-angiogenic therapy greatly extended the period of growth delay obtained.25,26,41 However, for this effect to be durable it is necessary that the anti-angiogenic drugs be maintained more or less indefinitely. If treatment is stopped for any reason, angiogenesis quickly resumes resulting in revascularization of tumour tissue and the resumption of tumour growth. 42 An alternative approach, and the one we favor, is to instead kill the endothelial cells in and around the viable rim that survive VDA treatment thus irreversibly preventing their ability to revascularize the tumour. Specifically, it was hypothesized that while endothelial cells in normal tissues are relatively resistant to the vascular disrupting effects of VDAs as a result of their dependancy on actin rather than tubulin for the maintenance of cell shape, exposure of such cells to the drug in the course of treatment might nevertheless induce changes that might be exploited in the development and/or optimization of effective combination therapies.
The spatial and temporal nature of radiation therapy makes it well suited for use in conjunction with VDA treatment. A number of such studies have been undertaken and encouraging results obtained in most cases.17,35–37,40,43–49 The order and timing in which the two therapies were used was important. Specifically, if radiation was given immediately after administration of a VDA there was little improvement in outcome.19,36,37,40,49,50 Explanations for this finding have generally focused on physiologic considerations and in particular, the possibility that the vessel shutdown induced by VDA treatment may produce transient hypoxia.19,49,50 As hypoxic cells show enhanced radiation resistance, any benefit that might result from prior treatment with a VDA will only be evident when this situation resolves. Thus Masunaga et al 49 have shown that there is an inverse temporal relationship between the degree of tumour control obtained with the hypoxic cytotoxin tirapazamine and radiation following administration of the VDA ZD6126. Tirapazamine was most effective when given within 1 hr of VDA treatment and had little activity if given after 24 hr. 49 Radiation, in contrast, gave little additional benefit at the 1 hr time point and was most effective when administered 24 hr after VDA treatment, particularly in the case of larger tumours. 49
Up to this point, the effect of VDA treatment on the intrinsic radiation sensitivity of normal and malignant cells has not been determined. This is an important variable that needs to be taken into consideration in order to achieve maximum benefit when VDAs are used in combination with radiation therapy in the treatment of solid tumours. In this regard, the results of the present study demonstrate that in addition to effects on cell shape and adherence, brief exposure of endothelial cells to ANG501, a novel stilbene VDA closely related to combretastatin A4, induces expression of a number of functionally important proteins including p21Wafl and the stress response genes HSP25, HSP70 and anti-B-crystallin. Although viability remains high, treated cells also transiently arrest in G2/M. As G2/M is generally the most radiation sensitive stage of the cell cycle27,28 it came as no surprise that endothelial cells treated with ANG501 also showed increased sensitivity to the cytotoxic effects of ionizing radiation. It was encouraging, however, that significant effects were evident at low, therapeutically useful, doses of radiation.
Although a reasonable possibility, it remains to be determined whether these various events are functionally linked. p21Wafl is a key regulator of cell cycle progression. Generally speaking, if present at low levels, it is sequestered by cyclin D upon mitogenic stimulation promoting the activation of cyclin D-dependent kinases 4 and 6, thereby facilitating the rapid movement of cells through the cell cycle. High levels of p21Wafl, in contrast, act to inhibit cyclin E-dependent kinase 2 resulting in cell cycle arrest. In mature endothelial cells, loss of one p21 allele resulted in decreased p21 protein expression and increased proliferative activity.
52
Apoptotic responses were also reduced while blood vessel formation in response to angiogenic stimuli
The mechanism by which ANG501 produces transient increases in the expression of p21Wafl is unclear. p21Wafl is controlled both transcriptionally by p53 and post-transcriptionally via proteosome dependent degradation. Previous studies have shown that various tubulin-binding anti-mitotic agents including paclitaxel, nocodazole, vinblastine, and colchicine 54 can also induce expression of p21Wafl. Taxol, has similarly been reported to induce p21Wafl in a dose- and time-dependent fashion. 55 In this instance, the effect appeared largely independent of p53 but involved activation of c-raf-1. 55 Taken together, these studies suggest that perturbation of tubulin polymerization/depolymerization may trigger the upregulation of p21Wafl expression.
The enhanced expression of certain heat shock proteins including HSP25, HSP70 and anti-B-crystallin following treatment of endothelial cells with ANG501 may also have important functional consequences. Heat shock proteins are induced in response to a broad range of stress-related stimuli including heat, radiation and infection. They function as molecular chaperones playing a key role in protein folding. 56 They also facilitate the generation of specific immune responses against both tumour antigens and infectious agents. 56 Importantly, heat shock proteins have been implicated in the control of cell cycling under both normal57,58 and stress-induced conditions. 59 Of potential relevance to the present study, elevated levels of HSP70 have been shown to reduce p21Wafl expression and ameliorate G2/M arrest in response to radiation, reducing the magnitude of radiation-induced cell death. 60
Tubulin destabilizing agents such as colchicine have been shown to induce HSP70 (HSP74) expression in the absence of elevated temperature. 61 Interestingly, the tubulin stabilizing agent Taxol did not produce similar induction even when cells were exposed to elevated temperatures. 61
It is evident from the studies described above, that other tubulin destabilizing agents such as colchicine produce changes in gene expression and cell cycling that are similar to those triggered by ANG501 treatment. Indeed, colchicine has been shown to induce G2/M arrest and function as an effective radiation sensitizer
Taken together, the results of the present study emphasize the need to consider the possible impact of VDA treatment not only on tumour physiology but also on intrinsic radiation sensitivity when developing or optimizing approaches in which these novel compounds are used in combination with radiation therapy in the treatment of solid tumours. The question of sequence and timing is likely to be particularly important in the context of fractionated radiation regimens. It is encouraging in this regard that non-cytotoxic doses of ANG501 can significantly enhance the sensitivity of endothelial cells to clinically relevant doses of radiation. Although the absolute increase in radiation-induced cell death achieved by addition of ANG501 is reasonably modest, if repeated throughout the course of a fractionated radiation regimen, then the impact on tumour survival would be dramatic.
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgements
These studies were supported by a pilot grant from the Women's Cancers Better Than Ever Program. The Arizona Center is the recipient of a Cancer Center Support Grant from the National Cancer Institute (CA23074).