
Abstract
Pro-oxidative stressors including cigarette smoke (CS) generate novel lipids with platelet-activated factor-receptor (PAF-R) agonistic activity mediate systemic immunosuppression, one of the most recognized events in promoting carcinogenesis. Our previous studies have established that these oxidized-PAF-R-agonists augment murine B16F10 melanoma tumor growth in a PAF-R-dependent manner because of its effects on host immunity. As CS generates PAF-R agonists, the current studies sought to determine the impact of PAF-R agonists on lung cancer growth and metastasis. Using the murine Lewis Lung Carcinoma (LLC1) model, we demonstrate that treatment of C57BL/6 mice with a PAF-R agonist augments tumor growth and lung metastasis in a PAF-R-dependent manner as these findings were not seen in PAF-R-deficient mice. Importantly, this effect was because of host rather than tumor cells PAF-R dependent as LLC1 cells do not express functional PAF-R. These findings indicate that experimental lung cancer progression can be modulated by the PAF system.
Keywords
Introduction
Lung cancer continues to present a dramatic clinical burden, accounting for nearly 14% of all new cancers diagnosed in the United States.1,2 Cigarette smoking (CS) alone contributes to more than two-thirds of lung carcinomas including small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC).3–5 Among all the critical signaling pathways involved, factors contributing to tumor escape mechanisms and host immunosuppression are emerging as important events in promoting tumor growth.6–9
Platelet-activating factor (PAF; 1-hexadecyl-2-acetyl-glycerophosphocholine) is a potent phospholipid-derived mediator that has been shown to mediate a wide range of pathologic responses from acute inflammation to delayed systemic immunosuppression.10–15 PAF agonists can be produced non-enzymatically in response to various pro-oxidative stressors via free-radical attacks on the sn2 position of cellular membrane, phosphocholine lipids.12–21 PAF exerts its effects via a seven-transmembrane G-protein-coupled receptor, the PAF-receptor (PAF-R), expressed on a variety of cells types.10,11,22,23
Several studies including ours have demonstrated that these oxidized PAF and PAF-like novel lipids mediate pro-oxidative stressor-induced systemic immunosuppression from ultraviolet B (UVB) and CS via a mechanism that requires host PAF-R-dependent enzyme, cyclooxygenase type 2 (COX-2), regulatory T cells (Tregs), and the cytokine interleukin 10
(IL-10).13–21 Of significance, we have recently demonstrated that this UVB/PAF-R agonist-mediated systemic immunosuppression augments the growth of experimental murine B16F10 melanoma tumors in a PAF-R-dependent manner. 19 Importantly, these effects were mediated via host rather than tumor cell PAF-R expression and mimicked by systemic administration of the metabolically stable PAF-R agonist, N-methyl carbamoyl PAF (CPAF). 19 In addition of having pronounced effects in the modulation of tumor growth, PAF/PAF-R signaling has been shown to play important roles in promoting angiogenesis and metastasis.24–31
Given that CS exposure remains one of the major causal factors in lung tumorigenesis and the fact that immunosuppression plays important role in promoting tumor growth as well as our findings that PAF-R agonists are critical in meditating systemic immunosuppression and inhibit host anti-tumor immunity via modulating Tregs, the current studies are designed to test if systemic PAF-R activation could modulate the growth and metastatic behavior of experimental lung cancer.
Material and Methods
Reagents
The RNA isolation, cDNA synthesis kits, and SYBR green quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) reagents were purchased from Qiagen (Maryland, USA). Murine PAF-R (catalog# PPM03062A), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) RT 2 primers were from SABiosciences (Valencia, CA). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO).
Cells and Mice
Murine Lewis Lung Carcinoma (LLC1) cells were purchased from ATCC (Manassas, VA). This cell line was established from the lung of a C57BL mouse bearing a tumor resulting from an implantation of primary LLC1. LLC1 cells were cultured in Dulbecco's modified eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 100 |Jg/ml penicillin and streptomycin. Murine B16F10 and human SK23mel melanoma cells were a kind gift from Dr. Christopher E. Touloukian (Department of Surgery, Indiana University School of Medicine [IUSM], Indianapolis, IN). PAF-R expressing B16-PAFR and human KBP cells were created and characterized by us previously.18–20,32 These cells were maintained in either DMEM or Roswell Park Memorial Institute medium (RPMI) media with above mentioned supplements. PAF-R expressing C57BL/6-wild-type (WT) mice were purchased from Charles River Laboratories. PAF-R-deficient (PAFR-KO) mice on a C57BL/6 background were a kind gift from Prof. Takao Shimizu (Department of Biochemistry, University of Tokyo, Tokyo, Japan). 33 These mice, aged 8-12 weeks, were housed under pathogen-free conditions and all procedures were approved by Institutional Animal Care and Use Committee (IACUC) at the IUSM.
In Vivo Tumor Growth and Metastasis Studies
A group of 12-14 WT and PAFR-KO mice were injected 0.2 × 10 6 LLC1 cells subcutaneously into right hind flanks. CPAF (250 ng/mouse in 100 μl PBS) or vehicle (100 μl PBS) were injected intraperitoneally (i.p.) as shown in Figure 1A. The optimum LLC1 cell number, 0.2 × 10 6 , was selected through pilot studies ± CPAF with different numbers of LLC1 cells in WT mice (n = 6 mice/group, data not shown). Tumor growth measured q2-3 days with digital caliper, and tumor volume was calculated (major circumference × minor circumference 2 /2). The various groups are being represented by arrows, pointing either upward or downward. The tails of these arrows are error bars representing the mean ± SE values of tumor volume.
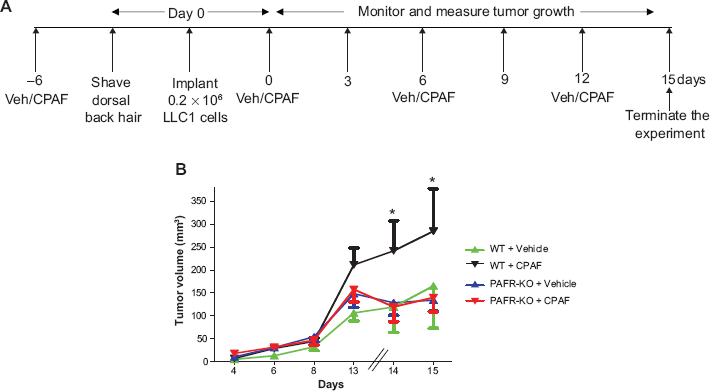
Effect of CPAF on the growth of LLC1 tumors in WT vs PAFR-KO mice. A) Schematic representation of the experimental protocol. B) A group of 12-14 WT and PAFR-KO mice were injected either with vehicle (100 μl PBS) or 250 ng of CPAF/mouse at day –6. At day 0, 0.2 × 10 6 LLC1 cells were implanted on the shaved dorsal hind flanks. CPAF treatments were repeated at day 0, 6 and 12. Tumor growth measured at various days with digital caliper. Data is represented as mean ± SE values of tumor volume over the period of time. *Denotes statistical significant differences (p ≤ 0.05) between CPAF-treated WT mice with other groups.
For spontaneous model of lung metastasis, at day 29, these WT/PAFR-KO mice were sacrificed and lung mets were counted. The lung tissues surrounding mets were formalin fixed, paraffin embedded (FFPE), and processed for the hematoxylin and eosin (H&E) staining (5 sections/met) for histology. These lung mets were confirmed by pathologist. The tumor cells showed an increased nuclear to cytoplasmic ratio and have hyperchromatic, clumped chromatin structure. The normal lung architecture was effaced with obliteration of the peribronchial alveoli.
Real-Time RT-PCR
PAF-R mRNA was analyzed in LLC1, B16F10, B16-PAFR, KBP, and SK23mel cells by qPCR and normalized with GAPDH similar to that described in Ref. 19 Briefly, total RNA was extracted from these cells using RNAeasy kit as per manual's protocol. Purified RNA was reverse transcribed using Super Script cDNA synthesis kit with random hexamers. cDNA was analyzed for the PAF-R gene using SYBR green-based, quantitative fluorescent PCR method. Fluorescence was detected with the StepOne Real-time PCR machine (Applied Biosystems, Foster City, CA). Quantification of each PCR product was normalized to GAPDH using the δδCt method.
Intracellular Calcium Ion Influx
Functional PAF-R in LLC1 cells was accessed by intracellular Ca2+ mobilization assay as described previously.19,20 In brief, LLC1 tumor cells, grown at optimum condition, were loaded with the Ca2+- sensitive fura-2-AM dye (4 μM in Hanks' balanced salt solution [HBSS]) at 37°C for 90 min. After that, cells were washed, resuspended in HBSS at room temperature, and CPAF (dissolved in 1% ethanol, adjusted to 1 μM) was added to an aliquot of these cells (1.0–1.5 × 10 6 LLC1 cells/2 ml). Similarly, LLC1 cells treated with 1 μM SLIGRL, protease-activated receptor (PAR) agonists (for which LLC1 cells express functional receptor), were used as a positive control. Fura-2-AM fluorescence was monitored in a Hitachi F-4010 spectrophotometer with excitation and emission wavelengths of 331 and 410 nm, respectively. The Ca2+ influx in suspensions was calculated as percentage of maximal peak calcium flux induced by 1 μM CPAF.
Cell Proliferation Assay
Cell proliferation was assessed by trypan blue exclusion assay using countess automated cell counter (Invitrogen, Eugene, OR). Briefly, LLC1 cells were plated at 5 × 10 5 cells/100 mm discs in triplicate and cultured overnight. These cells were treated either with vehicle (100 μl PBS in 1% ethanol) or 10 μM CPAF (dissolved in 100 μl PBS with 1% ethanol) and incubated for 24 and 48 hours. After each time points, cells were trypsinized, washed, resuspended in media and 100 μl of the media containing cells were stained with trypan blue and cells were counted with digital cell counter. Finally, cell numbers from each time points and treatment groups were normalized to 10 6 cells.
Statistical Analysis
Statistical analyses were conducted using Graph Pad Prism version 5. Student's t-tests and one-way analysis of variance (ANOVA) were used for comparing two or more groups. Statistical significance was defined as a P < 0.05.
Results and Discussion
Since CS exposure acts as a pro-oxidative stressor and is a major risk factor in lung cancer development and PAF-R agonists mediate CS-induced systemic immunosuppression, the current studies sought to determine the role of PAF/PAF-R signaling in lung cancer development and metastasis. As shown in Figure 1, exogenous administration of CPAF augments the growth of subcutaneously implanted LLC1 tumors in WT but not in PAFR-KO mice, which indicates that this effect is mediated via the host PAF-R. These results are consistent with our previous reports demonstrating that UVB/CPAF augments the growth of murine B16F10 tumors in a PAF-R-dependent manner. 19 These studies also indicate that PAF-R agonist-mediated systemic immunosuppression is not specific in augmenting the growth of experimental melanoma tumors; however, it can also affect the growth of lung cancer. Notably, pro-oxidative stressors other than CS and UVB, such as tert-butyl hydroperoxide and more clinically relevant chemotherapeutic drugs, have also been reported to generate oxidized lipids having PAF-R agonistic activity.34,35
Lung cancer metastasis, particularly in NSCLC accounts for >70% deaths. 36 Metastasis of lung carcinomas is a complex process that requires several stages from local tumor invasion to intra and extravasation and the formation of macrometastases. 36 Notably, PAF/PAF-R signaling has been shown to promote lung metastasis of melanoma and breast cancer cells directly by modulating various signaling cascades via tumoral PAF-R.27–31,37,38 We tested whether exogenous CPAF that augments PAF-R-dependent LLC1 tumor growth could modulate spontaneous LLC1 tumor cell metastasis to the lung. Notably, most tumor xenografts do not metastasize. We observed that CPAF-treated WT mice exhibited increased numbers of lung metastasis (Fig. 2A) compared to PAF-R-KO mice irepresentative picture of lung met histology is shown in Fig. 2B). These studies indicate that PAF-R agonists augment spontaneous lung metastasis in a PAF-R-dependent manner.
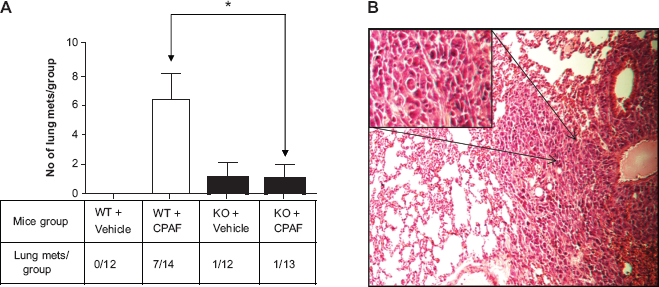
Effect of CPAF on LLC1 lung mets in WT vs PAFR-KO hosts. A) WT and PAFR-KO mice harboring LLC1 tumor cells were analyzed for spontaneous lung metastasis. After tumor measurements (shown in Fig. 1), mice were monitored for upto 29 days. At day 29, mice were sacrificed, lungs harvested and mets were counted. Data are represented as mean ± SE of total lung mets/group. *Denotes statistical significant difference (p ≤ 0.05) between CPAF-treated WT mice vs CPAF-treated PAFR-KO mice. B) Lung mets were paraffin embedded and sections stained with H&E. These mets were confirmed by Pathologist. A representative picture of lung tissue histology showing typical characteristics of mets is shown (arrows denote an area of lung met that has been highlighted in an inset).
Several studies have shown the direct effect of tumor cells expressing PAF-R in mediating PAF-R agonists induced effects.39–41 In contrast, our previous studies, via the use of genetically modified B16F10 cells stably expressing PAF-R, have shown that CPAF mediated enhanced growth of melanoma tumors was due to its direct effects on the host rather than tumor cell PAF-R as CPAF treatment exerted equal growth promoting effects on B16F10 tumors in WT mice implanted either with PAF-R-deficient or sufficient B16F10 cells. 19 However, CPAF treatment did not affect tumor growth in PAF-R-KO mice regardless of the PAF-R status of B16F10 cells. LLC1 cells do not express PAF-R mRNA as shown by qPCR (Fig. 3A). Calcium mobilization studies revealed that LLC1 tumor cells do not express functional PAF-Rs (Fig. 3B). Moreover, we did not observe increased growth of LLC1 cells in vitro by CPAF treatment (Fig. 3C).
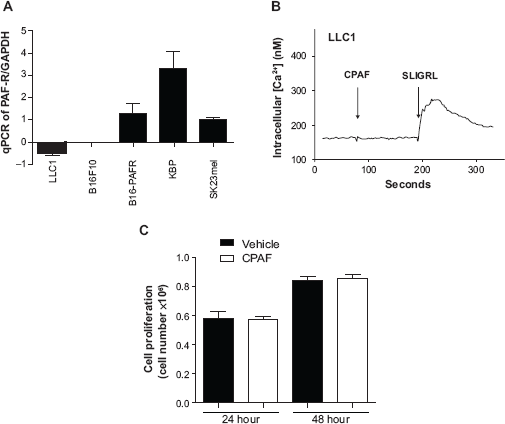
Functional characterization of LLC1 tumor cells. A) qPCR was performed to analyze the expression of the PAF-R in LLC1 tumor cells. PAF-R deficient murine B16F10 and ectopic PAF-R expressing B16-PAFR melanoma cells were used as negative and positive controls. In addition, ectopic and endogenous PAF-R expressing human KBP and sK23mel cells were used as positive controls. B) Intracellular calcium (Ca2+) assay was performed with LLC1 cells treated with 1 μM CPAF. LLC1 cells treated with 1 μM SLIGRL, a protease activated receptor (PAR) agonist was used as a positive control. C) Cell proliferation assay was performed in LLC1 tumor cells treated either with vehicle (0.1% ethanol) or CPAF and cultured for 24 and 48 hours. After each time points, cells were trypsinized, stained with trypan blue and counted by digital cell counter.
In summary, the current studies indicate that systemic PAF-R agonist augments the growth and spontaneous metastasis of lung tumors via effects on host PAF-R. These findings have clinical implications as they provide a novel pathway by which pro-oxidative stressors can promote lung cancer growth and metastasis via generating immunosuppressive lipid mediators having PAF-R agonistic activity.
Author Contributions
Conceived and designed the experiments: RPS and JBT. Analyzed the data: RPS, PCH, and JBT. Carried out the majority of experiments: PCH and RPS. Performed histological assessments and analysis: SR. Wrote the first draft of the manuscript: RPS. Contributed to the writing: JBT, RLK, and PCH. Agree with manuscript results and conclusions: PCH, SR, RLK, JBT, and RPS. Jointly developed the structure and arguments for the paper: RPS, JBT, and RLK. Made critical revisions and approved final version: PCH, SR, RLK, JBT, and RPS. Contributed reagents/materials/analysis tools: RPS, RLK, and JBT. All authors reviewed and approved the final manuscript.
Disclosures and Ethics
This paper was subject to independent, expert peer review by a minimum of two blind peer reviewers. All editorial decisions were made by the independent academic editor. All authors have provided signed confirmation of their compliance with ethical and legal obligations including (but not limited to) use of any copyrighted material, compliance with ICMJE authorship and competing interests disclosure guidelines and, where applicable, compliance with legal and ethical guidelines on human and animal research participants.