
Abstract
In this article, we have reviewed current literature regarding the regulation of hepatocellular carcinoma (HCC) by the interaction of malignant hepatocytes and their tissue environment through cytokine signaling, here represented by transforming growth factor-beta (TGF-β) signaling. We have discussed responses of TGF-β signaling in transition of hepatic stellate cells to myofibroblasts (MFBs), recruitment of tumor-associated macrophages (TAMs), and enrichment of tumor-associated endothelial cells (TECs). The malignant hepatocytes also secrete various factors such as platelet-derived growth factors (PDGFs), vascular endothelial growth factor (VEGF), and TGF-β. TGF-β, a super-family of cytokines, creates tumor microenvironment by interacting through other growth factors (epidermal growth factor receptor (EGFR), PDGF, fibroblast growth factor (FGF), hepatocyte growth factor (HGF), VEGF), cytokines and chemokines, and extracellular matrix (ECM) remodeling. Hence, the HCC tumor microenvironment may now be recognized as an important participant of tumor progression to act as potential target to systemic therapies compared to targeted therapies.
Introduction
Microenvironment is a system that consists of extracellular matrix (ECM) proteins, soluble factors, and signaling molecules such as cytokines and chemokines, along with a variety of cell types such as fibroblasts, immune cells, and endothelial cells. Under normal conditions, the microenvironment constitutes an important modulator of epithelium cell fate and a barrier to cell transformation.1,2 In cancer or in tumor progression, the microenvironment has drastic changes including the recruitment and the activation of stromal cells and the remodeling of ECM. Extensive evidence from genetics, genomics, and cell biology showed the established crosstalk between malignant hepatocyte and its cellular surrounding tumor microenvironment. The alterations at the genetic level in normal hepatocyte remodulate the expression of receptors, levels of growth factors, cytokines, and ECM components to guide the formation of malignant hepatocyte. Malignant hepatocyte displays complex interactions with tissue environment (consisting of biologically active cellular and biochemical components) leading toward tumor heterogeneity. The heterogeneity limits the efficacy of single drug treatment and provides insights to consider the entire tissue microenvironment as a biological target. Hence, it becomes necessary to explore the interactive and quite complex communication between the tumor and its microenvironment components to develop potential systemic therapies.
In hepatocellular carcinoma (HCC), malignant hepatocytes secrete various factors such as platelet-derived growth factors (PDGFs), vascular endothelial growth factor (VEGF), and transforming growth factor-beta (TGF-β), which interact with each other and also activate various surrounding cellular components such as fibroblasts (cancer-associated fibroblasts (CAFs) and hepatic stellate cells (HSCs)), 3 macrophages (tumor-associated macrophages (TAMs)), endothelial cells (tumor-associated endothelial cells (TECs)), and ECM remodeling. The activated cellular component in turn secretes plethora of factors (TGF-β1, epidermal growth factor (EGF), VEGF, PDGF, and basic fibroblast growth factor (FGF)) to promote HCC progression and invasion. In this review, we will analyze the multidirectional function of TGF-β, a super-family of cytokines in tumor microenvironment1,2 vis-a-vis cellular and non-cellular components and impact of systemic therapy on this interaction.
TGF-β Structure and Receptor
TGF-β is a pleiotropic cytokine that controls proliferation, cellular differentiation, adhesion, migration, apoptosis, and other functions.1,2,4 It has three isoforms (TGF-β1, TGF-β2, and TGF-β3) in mammals 1 and exists as two main receptors, called TGF-β receptor I or activin receptor-like kinase (ALK) (TpRI/ALK), and TGF-β receptor II (TβRII). The receptors also show variability: seven types of receptor I and five types of receptor II have been reported in humans. 4
Ligands such as bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), anti-Mullerian hormone (AMH), activin, nodal, and TGF-β's 5 bind to the extracellular domain of TβRII receptor, forms a ligand homodimer, and subsequently transactivates two TPRI/ALK5 dimers through phosphorylation of GS domain (which consists of a series of about 30 glycine-serine repeats) to form a stable heterotetrameric complex. 6 Consequently, TPRI/ALK5 facilitates phosphorylation of receptor-regulated SMADs (R-SMADs, namely SMAD1, SMAD2, SMAD3, SMAD5, SMAD8, and SMAD9). With TGF-β's, activins, nodals, and some GDFs SMAD2 and SMAD3 are phosphorylated, while BMPs, AMH, and a few other GDFs phosphorylate SMAD1, SMAD5, and SMAD9. The binding of R-SMAD to the type I receptor is mediated by a zinc double finger FYVE domain containing protein. Two proteins that mediate the TGF-β pathway include SARA (the SMAD anchor for receptor activation) and HGS (hepatocyte growth factor (HGF)-regulated tyrosine kinase substrate). SARA is present in an early endo-some which, by clathrin-mediated endocytosis, internalizes the receptor complex. 7 SARA recruits an R-SMAD. SARA permits the binding of R-SMAD to the L45 region of the Type I receptor. 8 SARA orients the R-SMAD such that serine residue on its C-terminus faces the catalytic region of the Type I receptor. The Type I receptor phosphorylates the serine residue of the R-SMAD. Phosphorylation induces a conformational change in the MH2 domain of the R-SMAD and its subsequent dissociation from the receptor complex and SARA. 9 Activated R-SMADs then heterodimerize with common-mediator SMADs (Co-SMADs, SMAD4), translocate to nucleus, and regulate downstream gene expression.10,11
This TGF-β pathway is tightly regulated, both positively 12 and negatively modulated by various ligands viz. extracellular antagonists, ligand-binding antagonists, regulation of receptor function, and inhibition by I-SMADs. Various ligands inhibit TGF-β signaling viz. chordin and noggin are antagonists of BMPs. They bind BMPs preventing the binding of the ligand to the receptor. 7 Members of the DAN family proteins such as Cerberus, DAN, and Gremlin antagonize GDF5, GDF6, and GDF7 ligands. Follistatin inhibits ligand activin. 13 Lefty is a regulator of TGF-β ligand. It acts by preventing the phosphorylation of R-SMADs. 14 Most importantly, drug-based antagonists have also been identified, such as SB431542, 15 which selectively inhibits ALK4, ALK5, and ALK7. Regulation of receptor function is coordinated through BMP and activin membrane bound inhibitor (BAMBI) with only extracellular domain, and no serine/threonine protein kinase domain binds to TGF-β receptor I and inhibits it. Similarly, FKBP12 prevents phosphorylation through binding to the GS domain of type I receptors. The most important negative regulation in the signaling pathway is the inhibition by I-SMAD (SMAD6 and SMAD7). SMAD7 prevents phosphorylation of R-SMADs and inhibits its binding to type I receptor. 16 SMAD6 binds SMAD4 preventing the binding of other R-SMADs with the Co-SMAD. In the nucleus, SMAD6 binds to the transcription factor Hoxc-8 and acts as an antagonist of transduction. SMAD6 could also bind to DNA directly and recruit histone deacetylase (HDAC) to inhibit the transduction. 17 The E3 ubiquitin-protein ligases SMURF1 and SMURF2 also regulate the levels of SMADs by accepting ubiquitin from an E2 conjugating enzyme and transferring it to the R-SMADs, which cause their ubiquitination and subsequent proteasomal degradation. SMURF1 binds to SMAD1 and SMAD5, whereas SMURF2 binds to SMAD1, SMAD2, SMAD3, SMAD6, and SMAD7. Hence, it enhances the inhibitory action of SMAD7 while reducing the transcriptional activities of SMAD2.
Non-SMAD Signaling Pathway
In addition to the SMAD pathway, TGF-β also activates other signaling cascades. For example, TGF-β activates the TGF-β-kinase 1 (TAK1) and Smad signaling; deletion of the same (TAK1) from hepatocytes results in spontaneous development of HCC, liver inflammation, and fibrosis. This is via TGF-β receptor-2 (Tgfbr2), which promotes HCC and liver ibrosis. 18 TGF-β activates the extracellular-signal-regulated kinase (ERK)-MAPK signaling pathway.19,20 Similarly, it also rapidly activates c-Jun N-terminal kinase (JNK) through MKK4 and p38 MAPK through MKK3/6 in several cell lines.21,22 In human prostate cancer cells, TAK1-SMAD7 inhibits the signaling pathway by initiating apoptosis; however, MKK3 and p38 MAPK help to activate the TAK1-p38 MAPK signaling pathway. 21 It is known that TGF-β is involved in activation of the phosphoinositide 3-kinase (PI3K)-AKT signaling pathway, and recent studies also indicate that TGF-β utilizes the mTOR pathway to regulate cell survival, metabolism, migration, and invasion through autophagy.23,24 Both the SMAD and non-SMAD signaling pathways determine the end results of the cellular response to TGF-β.
TGF-β interaction with cellular and non-cellular components of tissue microenvironment is given below.
Cellular Components
Transition of HSCs to Myofibroblasts (MFBs)
Transformed hepatocyte secretes TGF-β1, which promotes the transition of HSCs to MFBs through connective tissue growth factor (CTGF), thus establishing a crosstalk between MFBs and HCC through TGF-β1 in tumor development.25,26 MFBs as characterized by the expression of a-smooth muscle actin (a-SMA), fibroblast activation protein (FAP), fibroblast surface protein (FSP), and vimentin enhance the HCC progression through paracrine mechanisms.27,28 It has been documented that lysophosphatidic acid (LPA) induces HCC progression by recruiting peritumoral fibroblasts (PTFs) and promoting their trans-differentiation into MFBs. Hence, MFBs are the major component of the tumor microenvironment and play a crucial role in tumor-stromal interactions27–29 and promote liver cirrhosis, 30 tumor progression, invasion, and chemoresistance to clinical therapies.29,31,32 Importantly, they remodel the tissue microenvironment and facilitate the release of various growth factors and cytokines, and as a result magnify HCC growth.
The above evidences suggest that abrogation of TGF-β/SMAD signaling through various inhibitors may be of great importance. The use of LY2109761, which targets TGF-β receptor type I, can down-regulate CTGF production, which in turn interferes with the crosstalk between CAFs and HCC cells and hence inhibits stromal growth and metastasis. 26
Recruitment of TAMs
HCC cells recruit TAMs by secreting TGF-β, VEGF, PDGF, chemokine (C-C motif) ligand 2 (CCL2), and M-CSF, and by expressing glypican-3.28,33,34 These activated Kupffer cells or TAMs are mostly polarized toward the M2 phenotype, with increased levels of IL-10, arginase I, and IL-6 and decreased levels of IL-12, tumor necrosis factor (TNF), and proinflammatory cytokines such as nitric oxide (NO) and reactive oxygen species (ROS).35–38 This phenotype is characterized by low levels of antigen-presenting cells with a distinctive expression of Th-2 cytokines and interleukin-4 (IL-4), IL-10, and IL-13.39,40 When localized within peritumoral stroma, activated TAMs show a strong expression of human leukocyte antigen (HLA-DR), IL-6, IL-1p, and IL-23, whereas within the cancer niche, these TAMs exhibit a low expression of HLA-DR and IL-10. 41
The crosstalk between the damaged hepatocyte and TAMs has been documented in a diethylnitrosamine (DEN)-induced HCC mouse model system. DEN exposure promotes the release of IL-6 from Kupffer cells in response to IL-1a from damaged hepatocytes. Subsequently, IL-6 promotes an abnormal proliferation of the hepatocytes by triggering the signal transducer and activator of transcription 3 (STAT-3) and ERK pathways, which in turn control target genes involved in both cell proliferation and survival, thus contributing to HCC progression. 42 The TAM-derived cytokines and growth factors also contribute to induce immune suppression, which is essential for tumor growth. IL-10 and TNF-α, secreted by TAMs in an autocrine manner, stimulate B7-H1 expression on macrophages and impair CD8+ T cell activity, supporting tumor cells' immune escape. 43 Hence, above incidences suggest that an increased number of TAMs is correlated with the proliferation of tumor cells, angiogenesis, metastasis, and a poor prognosis.37,44
Enrichment of TECs
Tumor-associated endothelial cell (TEC), a major cell type in tumor microenvironment characterized through high expression of the endoglin marker (also known as CD105 or TGF-β receptor), 34 shows a rapid turnover rate, and enhanced mobility and migration compared to their normal part. 34 They contain several secretory organelles such as Weibel-Palade bodies, tissue plasminogen activator (tPA) organelles, and type-2 chemokine containing organelles. These organelles secrete tPA, cytokines IL-8 and IL-6, monocyte chemoattractant protein-1 (MCP-1), and growth-regulated oncogene-α (GRO-α).34,45,46 These cytokines enhance the process of migration and angiogenesis to provide invasiveness to the residual HCC. 30
Most importantly, TECs also stimulate HSCs to secrete both PDGF and VEGF. VEGF is one of the major growth factor responsible for angiogenesis in HCC. They are important for blood vessel formation and normal physiological processes such as thrombosis and inflammation, and also in tumor neoangiogenesis 47 through VEGF-A receptor. VEGF-A-AKT/mTOR and VEGF-NF-κB 48 signaling pathways have also been reported to activate VEGF expression 49 under proinflammatory conditions, one of the hallmarks of cancer progression.
Non-Cellular Components
TGF-β1
TGF-β1 growth factor is secreted in the ECM in a form of inactive complex called the large latent complex (LLC), formed through combination of small latent complex (TGF-β homodimer with precursor propeptide latency-associated peptide) and latent TGF-β binding protein. 50 The inactive LLC is turned on through matrix metalloproteinases (MMP-2 and MMP-9), 51 present in abundance in tumor environment. The activated TGF-β1 binds to TβRII and triggers TGF-β receptor-I (TpRI/ALK) phosphorylation to activate SMAD-2 and -3. TGF-β1 plays a dual role in HCC pathogenesis; in its tumor suppressor form (pre-malignant/normal state), it not only inhibits proliferation through activation of cyclin-dependent kinase inhibitors, suppression of c-Myc, and activation of apoptosis 52 in hepatocytes but also inhibits the stromal mitogens 53 and shows inhibitory action. However, in its oncogenic form, transition of TGF-β1 occurs through various mechanisms.53,54 Most commonly, HBx and HCV shifttumor-suppressive C-terminally phosphorylated Smad3 (pSmad3C) pathway to oncogenic linker-phosphorylated Smad3 (pSmad3L) pathway through activation of JNK.55,56 A recent study by Sohn et al showed that abrogation of the post-transcriptional regulation of c-Myc via methylation of a speciic single CpG site in the tristetrapolin (TTP) promoter presents a novel mechanism for the gain of selective resistance to the antiproliferative signaling of TGF-β1 in HCC. 57 In addition, increased levels of TGF-β1 in HCC tissues and perineoplastic stroma enhance liver ibrogenesis and hepatocarcinogenesis.51,58 Liver cirrhosis shows increased expression of TGF-β, which induces JNK-mediated hepatic stellate cell proliferation and CTGF (CCN2)-mediated ibrogenesis 59 and collagen production.60,61 Kluwe et al showed that pan-JNK inhibitors inhibit PDGF and TGF-β signaling, and prevent human HSC proliferation. 62 Hence, the above evidences prove the concept that tumor microenvironment affects the liver fibrogenesis and hepatocarcinogenesis.
EGF Receptor (EGFR)
Ligand-mediated EGFR activation and subsequent c-Src phosphorylation has also been observed in undifferentiated hepatocyte and liver tumor. A recent study establishes the fact that TGF-β-mediated activation of AKT is facilitated via EGFR ligand secretion. Hence, levels of EGFR deine the cell fate.59,63 Additionally, it has also been reported that once hepatocytes have undergone epithelial-mesenchymal transition (EMT), they acquire mesenchymal phenotype and activate autocrine loop of TGF-β, up-regulate TGF-β mediated oncogenic miR-181b expression, miR-23a, 27a, and 24, and enhance EGFR expression. Simultaneously, all these pathways confer resistance to apoptosis and promote carcinogenesis.64,65
PDGF
The PDGF secretion transforms HSCs (characterized through PDGF receptors during liver injury) into MFBs, and promotes ibrogenesis and proliferation which may lead to liver cirrhosis–-a prerequisite of carcinogenesis. This has been supported by Campbell et al. They showed that over-expression of PDGF-C in the liver of the transgenic mouse results in a number of proibrotic pathways in HSCs that lead to proliferation, tissue ibrosis, and subsequent development of HCC mediated through the activation of the ERK-1/-2 and PKB/AKT signaling pathways.66–68 Other than HSCs, PDGF is also secreted by HCC (malignant hepatocyte), and tumor endothelial cells activate transition of HSCs to MFBs to produce both PDGF and VEGF to support neovascularization. 69 The above evidences show how all cellular components of tissue microenvironment induce the activation of HSCs into MFBs, which may lead to liver cirrhosis–-a prerequisite of carcinogenesis.
The synergistic action of TGF-β and PDGF is well established. During acute liver injury, TGF-β and PDGF synergistically promote controlled collagen synthesis. Deposition of collagen occurs through dual phosphorylation of pSmad2L/C and pSmad3L/C at linker and carboxyl-terminal region pathways in the activated HSCs. However, unlimited ECM deposition is controlled by TGF-β-induced Smad7, which negatively regulates the ibrogenic TGF-β signaling.
On the other hand, in case of chronic liver injury a different pathway is followed. In the absence of Smad7 induction, both TGF-β and PDGF lead to sustained constitutive ibrogenesis through pSmad2L/C and mitogenic pSmad3L signals in MFBs, which eventually develop into accelerated liver ibrosis–-a major risk factor for carcinogenesis. 70 TGF-P1 also facilitates the EMT process through the activation of the PDGF signaling pathway. 71 However, only in vitro evidences support the concept whereas in vivo evidences are lacking. Upon EMT, TGF-β induces the secretion and autocrine regulation of PDGF through up-regulation of PDGF-A, and both PDGF receptors as proven through loss-of-function analyses in differentiated p19 (ARF) null hepatocytes. 72 PDGF regulates two important mechanisms: (i) activation of phosphatidylinositol-3 kinase and (ii) simultaneous release of β-catenin from its inhibitory complex to accumulate in nucleus leading to EMT. Hence, PDGF plays a role as an essential link to TGF-β signaling-mediated nuclear β-catenin accumulation in HCC progression as evaluated constitutively in expressing active β-catenin or its negative regulator axin hepatocytes. Fischer et al contradicted the hypothesis. They demonstrated that active β-catenin fails to accelerate proliferation and causes growth arrest. Simultaneous expression of the CDK inhibitor p16 (INK4A) with the nuclear accumulation of β-catenin induced downstream target genes cyclin D1 and c-Myc. Additionally, active β-catenin protects malignant hepatocytes against cellular death and provides stem cell-like property. Hence, TGF-β-mediated tumor progression is by induction of PDGF signaling and activation of β-catenin.
The above evidence suggests that regulation of ECM accumulation by TGF-β and PDGF signals involves different mechanisms in acute/chronic liver injuries and HCC even though HSCs are the principal effector in all the cases. As a result of chronic liver damage/HCC, HSCs undergo progressive activation to become MFB-like cells.
FGF
FGFs are growth factors that are involved in tissue regeneration, wound healing, and angiogenesis.73,74 However, aberrant expression of FGFs has been reported in HCC, and it has been found to promote HCC and endothelial cell proliferation through the activation of downstream ERK and AKT pathways. Chen et al showed comparative positive expression rate of TGF-β1 [84.1% (106/126) in hepatic tumors and 64.3% (81/126) in peritumoral liver tissues] and FGFR4 [74.6% (94/126) in hepatic tumors and 57.1% (72/126) in peritumoral liver tissues] in hepatic tumors and peritumoral liver tissues. 75 The long-term exposure to TGF-β in paracrine way induces isoform switching of FGFR and sensitizes the cells to FGF-2. Addition of FGF-2 to TGF-β-treated cells irst perturbs epithelial myoibroblasts transition (EMyoT) by reactivation of the MEK-ERK pathway and then enhances EMT through the formation of MEK–ERK-dependent complexes of the transcription factor 8EF1/ZEB1 with the transcriptional corepressor CTBP1. Consequently, the enhanced EMT as a result of combined TGF-β and FGF-2 stimulation promotes the invasion of cancer cells. Thus, TGF-β and FGF-2 cooperate with each other and regulate EMT during HCC progression. 76 In addition to EMT, FGF also regulates TGF-β signaling and endothelial-to-mesenchymal transition (Endo-MT) via control of let-7 miRNA expression. Disruption of baseline FGF signaling to the endothelium leads to a dramatic reduction in let-7 miRNA levels that, in turn, increases expression of TGF-β ligands and receptors and activation of TGF-β signaling, leading to Endo-MT. 77
HGF
HGF expressed through HSCs, MFBs, or CAFs acts as a mediator of tumor-stromal interactions to increase the proliferation and invasion of HCC cells. 78 HGF also inhibits collagen I and IV synthesis in HSCs by miRNA-29 induction, and collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. 79 Although it has been reported that HGF can act synergistically or antagonistically with TGF-β signaling, molecular mechanism of their crosstalk remains unknown. However, Mori et al reported that both HGF and TGF-β transmit the signals through JNK-mediated R-SMADs phosphorylation at linker regions as validated by JNK inhibitor SP600125. 80 Hence, up-regulation of HGF and down-regulation of TGF-β controls the anti- or profibrogenic response of HSC, respectively.
VEGFs
VEGF is one of the major growth factors responsible for angiogenesis in HCC. They are important for blood vessel formation and normal physiological processes such as thrombosis and inflammation, and also in tumor neoangiogenesis 47 through VEGF-A receptor. VEGF-A–AKT/mTOR and VEGF–NF-κB 48 signaling pathways have also been reported to activate VEGF expression 49 under proinflammatory conditions, one of the hallmarks of cancer progression.
Cytokines and Chemokines
Activated HSCs/MFBs secrete chemokines, such as MCP-1 (also known as CCL2), for chemoattraction of monocytes and contribute to the inflammatory infiltration of activated Kupffer cells or TAMs. 71 In turn, TAMs also secrete many cytokines, chemokines, and growth factors, which support HCC progression, and these TAM-derived signals are regulated at various levels and interact with tumor cells. Moreover, TAMs support angiogenesis, metastasis, and cancer progression. Therefore, the complex network of tumor–stroma crosstalk could be a promising source of therapy for HCC.
ECM Remodeling
HSCs converted to MFBs (activated HSCs) are also involved in ECM remodeling through increased synthesis of collagen types I and III and differential expression of MMPs and TIMPs. Mice with MMP-9 mutations inhibit fibrogenesis and results in decreased portal and periportal accumulation of collagen. MMP-9 mutations also suppress trans-differentiation of HSCs to the MFB-like phenotype in vitro and in vivo. 81 Both TGF-β1 and MMPs reciprocally activate each other and modulate ECM to promote migration and invasion of HCC cells. 64 MiR-181b, which is up-regulated by TGF-β1, up-regulates MMP-2 and -9 and promotes migration and invasion of HCC cells. 64 Communally, MMP-2, -9, and -14 also activate TGF-β1 secreted from the malignant hepatocyte, a key modulator of EMT in HCC. 65 MMPs are also associated with various pathways; high expression of MMP-9 is associated with activation of PI3K/PTEN/AKT/mTOR pathways82,83 and inhibition of apoptosis through MMP-7-mediated cleavage of Fas ligand in human HCC cells. 84
Activated TGF-β1 also triggers invasiveness and motility through laminin-5 (Ln-5; a cell adhesion glycoprotein that belongs to the laminin family and forms a mesh-like structure to resist the tensile forces in the basal lamina) by inducing the expression of transmembrane integrin receptor a3P1. 85 In HCC, Ln-5, together with TGF-β1, promotes EMT by over-expressing Snail and Slug and down-regulating E-cadherin, followed by translocation of β-catenin into the nucleus.85,86 Gefitinib, a specific inhibitor of EGFR, inhibits phosphorylation of the receptor and of the molecular pathways AKT and ERK1/2 on HCC cells in vitro and hence suppresses HCC growth. However, the presence of Ln-5 in HCC antagonizes gefitinib's efficacy in a dose-dependent manner, suggesting a potential drug (gefitinib) failure in Ln-5 positive HCC cases. 87
Moreover, Ln-5 is not detected in normal liver, but it is expressed in HCC nodules, and a high expression is associated with a more proliferative and metastatic phenotype of HCC. The presence of the Ln-5 γ2 chain in metastatic HCC is also correlated with poor prognosis and survival.88,89
TGF-β family members are the critical regulators of EMT in hepatocytes regulated through multiple pathways. They induce activation or inactivation of the small GTPases and the phosphorylation of Par6, which might be critical events leading to EMT. 90 Meyer et al and other groups reported that TGF-β1 down-regulates E-cadherin, zona occludens and up-regulates mesenchymal markers such as Snail, vimentin, and N-cadherin. 91
Systemic Therapy
A better understanding of this complex network of interactions between tumor cells and their milieu could provide new insight into novel targets for HCC treatment. The HCC tumor microenvironment may now be recognized as an important participant of tumor progression to act as potential target to systemic therapies compared to targeted therapies. Presently, systemic therapeutic options in the locally advanced or metastatic setting are limited to sorafenib, an oral multi-kinase inhibitor targeting Raf kinase, VEGF, and PDGF receptor tyrosine kinase signaling. 92 Sorafenib has resulted in a significant overall survival of patients with advanced HCC.92,93 Cetuximab, a monoclonal antibody against the EGFR, failed to show any significant activity against HCC in a phase II study. 91 Clinical trials using small molecules targeting EGFR, such as erlotinib, gefitinib, and lapatinib, were also ineffective against HCC.94–96 Various other multi-kinase inhibitors targeting VEGFR, such as sunitinib, axitinib, linifanib, pazopanib, vandetanib, cediranib, and regorafenib, as well as monoclonal antibodies like bevacizumab, are also under clinical trial to treat HCC. 97 However, many phase I/II clinical trial with multi-targeted agent, namely dovitinib, which targets VEGFR, PDGFR, and FGFR 19 and HDAC inhibitors (targeting at epigenetic level), CHR2845, and PXD101 (belinostat) 20 may inhibit TGF-β induced I-SMAD recruitment of HDAC to inhibit transduction against cancer-associated inflammation in HCC are currently underway. Some efforts are also being carried out with combination of transarterial chemoembolization (TACE) and anti-angiogenic agents such as sorafenib, but failed to observe any survival beneits. 22
The above evidences show the importance of the interaction of TGF-beta with cellular and non-cellular components of the tumor microenvironment (Fig. 1). Hence, the tumor microenvironment changes dynamically and consequently affects HCC initiation, progression, and metastasis. Therefore, it is necessary to explore the tumor microenvironment more thoroughly to understand the biological and molecular interactions between cellular and non-cellular components vis-a-vis malignant hepatocytes, which will help us in development of new therapeutics.
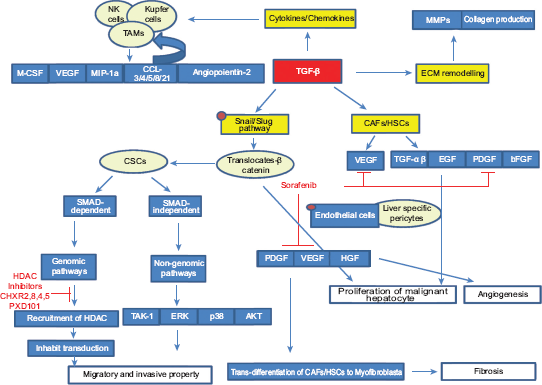
Tumor progression through interaction between CAFs and malignant hepatocyte. Cancer cells secrete TGF-β to activate fibroblasts which, in turn, produce a series of growth factors and cytokines that sustain tumor progression by promoting ECM remodeling, cell proliferation, fibrosis angiogenesis, and migration and invasive property.
Author Contributions
Conceived and designed the experiments: DKG and NS. Analyzed the data: NS. Wrote the irst draft of the manuscript: NS. Contributed to the writing of the manuscript: DKS. Agree with manuscript results and conclusions: DKG, NS and DKS. Jointly developed the structure and arguments for the paper: NS and DKS. Made critical revisions and approved final version: DKG. All authors reviewed and approved of the inal manuscript.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with iCMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.