
Abstract
Objective
Carbonic anhydrase (CA) II deficiency is a rare autosomal recessive disorder caused by mutation in the CA II gene that leads to osteopetrosis, renal tubular acidosis (RTA), and cerebral calcification. Our aim is to present a patient with the classic triad of CA II deficiency syndrome to enhance the awareness about this rare syndrome.
Methods
We describe the clinical and radiological findings of a Saudi woman patient with CA II deficiency syndrome.
Results
A Saudi woman in her 20s presented to our hospital for evaluation of increased bone density. She was known to have delayed developmental milestone with growth retardation and poor scholastic performance. She had multiple fragile fractures started at the age of 15 involving the lower extremities. A physical examination revealed dysmorphic features and intellectual disability with intelligence quotient (IQ) of 36. The initial blood workup showed a picture of distal RTA with hypokalemia, and the radiological imaging confirmed the presence of osteopetrosis and multiple kidney stones. The combination of osteopetrosis with RTA raised the possibility of CA II deficiency. Therefore, computed tomography (CT) of the brain was done and showed intracranial calcification involving the basal ganglia. She was started on potassium chloride and sodium bicarbonate. In addition, she underwent right-sided percutaneous nephrolithotripsy. Her DNA analysis came to show a sequence variant c.232+1G>A, which was detected in both of the CA II genes (homozygous).
Conclusion
Early recognition of the disease is a key, as an early appropriate treatment institution is essential in order to prevent further complications.
Case Report
A Saudi woman in her 20s presented to our hospital for evaluation of increased bone density. She is known to have delayed developmental milestone with growth retardation and poor scholastic performance. She was born to healthy consanguineous parents (both were from the same tribe, however, they were not close relatives) and was a product of full-term normal delivery. She had multiple fragile bone fractures that started at the age of 15 involving the lower extremities; one of them required open reduction and internal fixation for the left tibia. Six months prior to her presentation, she developed a sudden generalized weakness and was found to have severe hypokalemia of 1.4 mmol/L. There were no similar illnesses in the family.
A physical examination revealed dysmorphic features with a prominent forehead, prominent cheeks, micrognathia, teeth malalignment, and short digits in relative to arms and legs. Her height and weight were 132 cm and 42.7 kg, respectively. She had fluent nasal speech. Her intelligence quotient (IQ) by Wechsler Adult Intelligence Scale was 36. She had well-developed secondary sexual characteristics. A central nervous system examination revealed no neurological deficit. She had normal cranial nerves with normal sensory and motor systems. Cerebellar signs were within normal limits. Cardiovascular, chest, and abdominal examinations were unrevealing.
Laboratory investigations showed hypokalemia of 3 mmol/L (3.6–5.2) and a finding of distal renal tubular acidosis (RTA) with a pH of 7.28, HCO3 of 16.2 with hyperchloremia of 110 mmol/L (98–106), Na 141 mmol/L (135–145), and a urine pH of 6.5. Also, she had vitamin D deficiency of 40 nmol/L (50–250), hypocalcemia of 2.02 mmol/L (2.3–2.64), albumin 33 g/L (34–50), PO4 1.04 mmol/L (0.81–1.58), PTH 4.83 pmol/L (1.6–6.9), and creatinine 69 μmol/L (53–88). Her thyroid function test showed elevated TSH of 6.05 mIU/L (0.27–4.2), normal FT4 12.5 pmol/L (12–22), and normal FT3 3.69 pmol/L (2.8–7.1). Thyroperoxidase antibodies were elevated. She had normal CBC and liver enzymes.
Skeletal survey showed increased bone density (Fig. 1). Bone mineral density showed lumbar spine T-score of +5.4 with a Z-score of +6.1 and total hip of right femur T-score of +2.7 with a Z-score of +3.3. Kidney and urinary bladder X-rays, and renal ultrasound revealed right hydronephrosis with multiple bilateral renal calculi with the largest one measuring 1.6 cm in the upper pole of the right kidney (Fig. 2). The combination of osteopetrosis with RTA raised the possibility of carbonic anhydrase (CA) II deficiency. Therefore, computerized tomography of the brain was done and showed extensive symmetric intracranial calcification involving the subcortical fibers, basal ganglia, both thalamic and subthalamic nuclei, and both cerebellar hemispheres (Fig. 3).
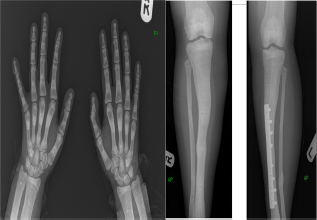
Skeletal survey showing generalized increased bone density.

An ultrasound of right kidney showed multiple renal stones.
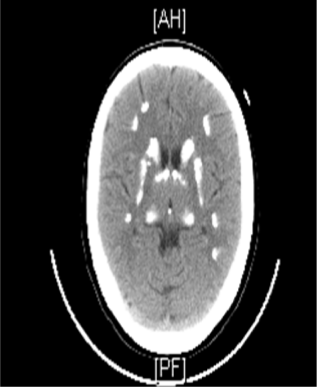
Noncontrasted computed tomography (CT) scan of the brain showing extensive symmetric intracranial calcification involving the basal ganglia.
She was started on potassium chloride, sodium bicarbonate, oral calcium, and vitamin D supplementation. She underwent right-sided percutaneous nephrolithotripsy. Her DNA analysis for CA II deficiency showed a sequence variant c.232+1G>A, which was detected in both of the CA II genes (homozygous). Thus, CA II deficiency with most of its manifestations was demonstrated in this case.
Discussion
CAs are called metalloenzymes as the active site contains zinc ion that helps catalyzing the reversible conversion of carbon dioxide to bicarbonate and releasing proton to maintain acid–base balance in blood and other tissues. 1 CA II is one of the most catalytically active CA isozymes, 2 with the widest tissue distribution being found in bone, kidney, brain, and erythrocytes. 3 It plays a major role in renal regulation of acid/ base homeostasis and osteoclast-mediated bone resorption, and its deficiency gives rise to a rare syndrome of osteopetrosis, RTA, and cerebral calcification. 4 The CA II deficiency syndrome is inherited in an autosomal recessive fashion and found most frequently in the Middle East and Mediterranean regions. 3
Deficiency of CA II impairs production of H
The gene for CA II is mapped to chromosome 8q22. 14 Although CA II deficiency has been reported in families from various ethnic origins (Belgian, American, French, German, Italian, Hispanic, Japanese), it is particularly common in the Middle East and more than 70% of the cases have been described from the Arabian Peninsula. 15 Patients of Arabic origin have a unique splice junction mutation at the junction of exon 2–intron 2 of the CA II gene (c.232+1G>A), 16 which is identical to our patient. Clinically, Arabic patients have a very severe phenotype and are distinguished from the American and Belgian patients (H107Y mutations) by the presence of severe cognitive impairment and relative infrequency of skeletal fractures. 17 This was attributed to the residual activity of the His-107–>Tyr mutant enzyme, which may explain the absence of intellectual disability and the relatively mild phenotype of CA II deficiency in the affected members.12,18
There is no established medical treatment for CA II deficiency, and the therapy is restricted to symptomatic correction of metabolic acidosis and hypokalemia. There is a well-known role of bone marrow transplantation (BMT) in autosomal recessive malignant osteopetrosis, a more aggressive form of osteopetrosis with potential complication of bone marrow failure. BMT can be curative in this condition, with almost an 80% five-year disease-free survival when the donor is HLA identical. However, the role of BMT in CA II deficiency is less clear and need careful consideration in terms of risks and benefits. This is mainly because some kindreds have a mild phenotype or the diagnosis may be delayed, and the patient may have irreparable neurologic damage. 3 The available data suggested that the allogeneic BMT in case of CA II deficiency reverses the osteopetrosis and retards the development of cerebral calcification, but it appears to have little or no effect on the RTA.3,19,20
Conclusion
Early recognition of the disease is a key, as an early treatment with bicarbonate for the systemic acidosis associated with RTA along with potassium supplementation is essential in order to prevent further complications. The phenotype of CA II deficiency is variable. In those with benign phenotype and relatively normal lives, the treatment is usually symptomatic as the risk of BMT in such patients outweighs the benefit. However, for those with severe damaging phenotype with severe skeletal abnormalities, BMT can offer a hope for improvement. 3
Author Contributions
Provided clinical care to the patient: ONA, OMA, SMS. Wrote the first draft of the manuscript: ONA. Contributed to the writing of the manuscript: ONA, OMA, SMS. Agree with manuscript results and conclusions: ONA, OMA, SMS. Jointly developed the structure and arguments for the paper: ONA, OMA, SMS. Made critical revisions and approved final version: ONA, OMA, SMS. All authors reviewed and approved of the final manuscript.