
Abstract
The objective of the present study was to validate prognostic gene signature for estrogen receptor alpha-positive (ERα+) and lymph node (+) breast cancer for improved selection of patients for adjuvant therapy In our previous study, we identified a group of seven genes (
Keywords
Introduction
Breast cancer is a heterogeneous disease, 1 making it an ideal disease to study using microarrays since different expression patterns can be identified within distinct tumor groups. An increased understanding of the pathogenesis of breast cancer is imperative in the pursuit of innovative therapies for treatment and/or prognosis of patients. Gene expression studies based on microarray have been used extensively by cancer researchers to profile cancer subsets, predict patients’ outcome, and identify genes of clinical relevance.2–5
Breast cancer is no longer a single disease, but it is a heterogeneous disease consisting of different subtypes on the molecular and histopathological levels with different prognostic and therapeutic outcomes.6–9 Gene expression profiling has classified breast cancer into five biologically distinct intrinsic subtypes: luminal A, luminal B, human epidermal growth factor receptor 2 (HER2+), basal-like, and normal-like.6–9 The luminal A subtype is estrogen receptor alpha positive (ERα+), progesterone receptor positive (PR+), and HER2 (–); luminal B subtype is ERα+, PR+, and HER+, and luminal B is associated with a relatively worse outcome. Both HER2 (+) and basal-like (ER-, PR-, and HER-) breast cancers have poor outcomes. Parker et al.
9
developed an efficient classifier, called PAM50, to distinguish these five intrinsic subtypes using the expression of 50 “classifier genes.” In a more recent study, a large breast cancer patient cohort (
An important part of the diagnostic workup of all the breast cancer patients is the determination of the ERα status of the tumor. Clinically, an ERα (+) status is associated with improved prognosis, lower risk of relapse, and better overall survival, 13 and that are key aspects for making decisions for endocrine therapy with antiestrogens. A major problem in clinical oncology is to distinguish the patients who are likely to present a relapse of the disease from those with a favorable prognosis. In recent years, it has been realized that apart from ERα, other factors are also important in deciding the therapeutic strategies of the patient. These include histological markers such as grade, tumor size, lymph node involvement, PR, and HER2 receptor status. Each of these has modest positive predictive value (30%-60%).14–17 Moreover, the current histological classifications of breast cancer do not fully represent the diverse clinical outcome of the disease. Recent approaches for patient management, which utilize histological markers in conjunction with online statistical algorithms such as “Nottingham Prognostic Index” and “Adjuvant! Online”, fail to predict the course of the disease in a significant number of breast cancer patients.18,19 Women with node (+), ERα (+), and HER2 (–) often receive adjuvant treatment with chemotherapy and hormonal therapy. Nevertheless, few patients eventually experience a recurrence. Thus, new tools are needed to allow improved definition of a risk of recurrence. If it were possible to predict cancer recurrence following standard therapy, these patients could be targeted for alternative treatment strategies.
Recently, we published gene expression profile of breast tumors and identified seven genes
Materials and Methods
Patients and breast cancer tissues
Human cells and specimens were obtained for previous research, 20 deidentified, and reused for this study. This research was therefore exempt from the requirement for ethics committee approval under US regulations §46.101(b)(4). The research was conducted in accordance with the standards in the Declaration of Helsinki. Breast tumor samples were obtained from patients undergoing surgery after informed written consent (Apollo Hospital). The excised tumor specimens were immediately preserved in RNA-later (Life Technologies) and stored at 4 °C until shipment. All tumor samples utilized in this study were invasive ductal carcinoma (Supplementary Table 1). All the histopathologi-cal information used in the analysis was directly documented from the original pathology reports. Grading, tumor type, ERα status, PR status, and HER2 status had been routinely recorded at the Apollo Hospital.
Public microarray data sets
Due to limited availability of clinical information in our data set, we used independent data sets to assess the predictive ability of our gene signatures, which will give us additional confidence in clinical validity. The data sets with Gene Expression Omnibus (GEO) accession numbers GSE2740, GSE1992, and GSE2607 generated using Agilent microarray platform have been previously described.21–23 A total of 141 patient samples in GSE2740, 152 patient samples in GSE1992, and 102 patient samples in GSE2607 were used. A total of 395 patient samples with histopathological information are available in Supplementary Table 2. Patient samples with missing survival information were omitted; hence, a total of 340 samples were used for further analysis (Table 1). The clinical data were extracted from the gene expression data files downloaded from GEO. The ERα status, nodal status, survival data, and gene expression data were used for Kaplan-Meier survival curve and Cox multivariate analysis.
The range of expression for seven genes and a median value used to stratify patient samples from public data sets into the groups of high expression and low expression.
The relation between mRNA levels and clinicopathological parameters.

Statistical analysis
To visualize gene expression values using heat maps, the values for each probe were centered by subtracting the mean expression value across patients. No gene-specific scaling (standardization) was performed, and, thus, information about the relative signal strength between probes was retained. The color tone in the heat maps was calibrated so that saturated red and saturated green were reached at values equal to 3.5-fold the standard deviation of the expression values of the entire matrix. Red and green reflect high- and low-expression levels (log 2-transformed scale), respectively.
The gene expression data from 340 patient samples were dichotomized according to the median (cutoffvalue 0.0) of the complete cohort. The expression data higher than the median were grouped into the “high-expression” group, and the expression values lesser than the median were grouped into the “low-expression” group (Table 1). Mann-Whitney
Results
Relationship between mRNA levels of seven genes and clinicopathological parameters
Initially, we sought to corroborate the findings of our recent study.
20
We correlated the mRNA expression of these seven genes with clinicopathological parameters. In our previous study,
20
we had performed reverse transcription quantitative polymerase chain reaction (RT-qPCR) analyses of 76 tumor specimens and showed that the expression of aforementioned seven genes was significantly associated with ERα (+) tumors (
Meta-analysis of seven genes deregulated in ERα (+) breast tumors
Next, we sought to correlate the mRNA expression of these seven genes with long-term survival data. Since we did not have an adequate number of clinical samples, we utilized published data sets to validate our predictive gene set. Further use of independent data sets to assess the predictive ability of our gene signatures will give us additional confidence in clinical validity. Accordingly, gene expression data were obtained from three independent public data sets (

Dendrogram of 340 breast cancer samples from public data sets. Unsupervised, hierarchical, uncentered Pearson distance (co-relation) clustering was performed to classify the seven genes into homogeneous clusters.
High mRNA expressions of GATA3, NTN4 , and MLPH are associated with longer RFS in ERα (+) breast tumors
The gene expression values from the public data sets were dichotomized according to the median of the complete cohort, and expression data higher than the median were grouped into the high-expression group, and the expression values lesser than the median were grouped into the low-expression group (Table 1). Univariate analysis on ERα (+) test data sets (

Kaplan-Meier survival curve using high and low mRNA expression among ERα (+) breast tumors from public data sets (
The univariate and multivariate analysis in relation to RFS among 195 ERα (+) breast cancer patient samples from public data sets.

High mRNA expressions of GATA3, SLC7A8 , and MLPH are associated with longer RFS in ERα (+) and node (+) breast tumors
Having studied the prognostic significance of the seven dysregulated genes in ERα (+) patients, we next sought to perform our analysis with the clinically important issue of the metastatic spread of the tumor. The determination of the extent of lymph node involvement in primary breast cancer is the single most important risk factor in disease outcome. Accordingly, we next investigated the correlation of these seven dysregulated genes with ERα (+) and node (+) cohort (
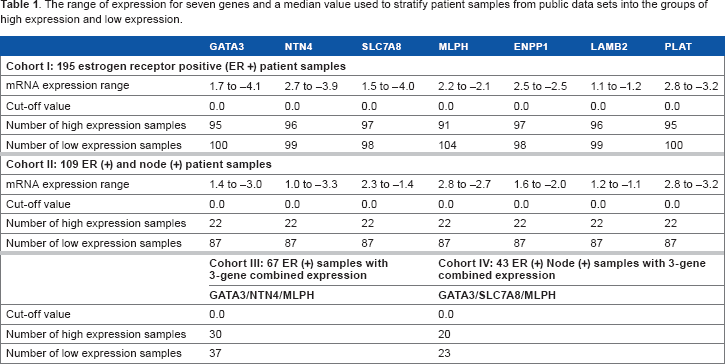
Univariate and multivariate analysis in relation to RFS among 109 ERα (+) and node (+) breast cancer patient samples from public data sets.
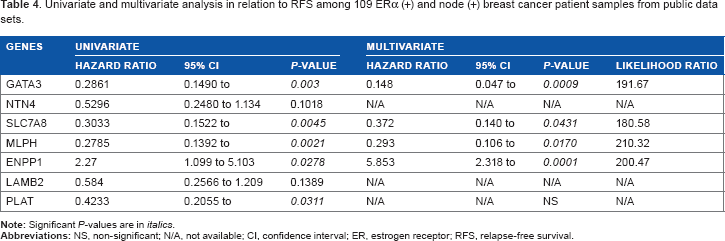
Univariate and multivariate analysis in relation to RFS among 84 ERα (+) and node (–) breast cancer patient samples from public data sets.
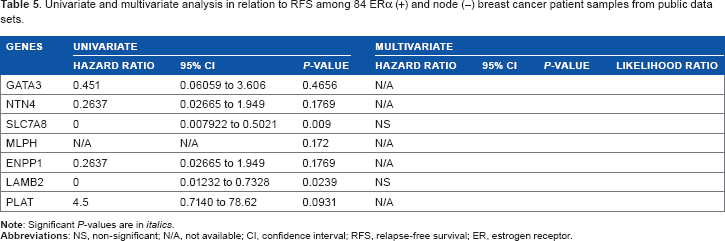
Elevated expression of three-gene signature improves RFS
In our previous analyses, we had ascertained the predictive power of “individual” genes. We next sought to determine the “combined” predictive power of the three-gene signature. Accordingly, patients who expressed high mRNA levels of “all” three genes were studied against those who expressed low mRNA levels of all three genes with the cutoff value of zero. Initially, we looked into the 195 samples of ERα (+) cohort. In multivariate analysis, the three genes
Univariate and multivariate analysis of three-gene signature in relation to RFS among ERα cohort (π = 67), and ERα (+) and node (+) samples (
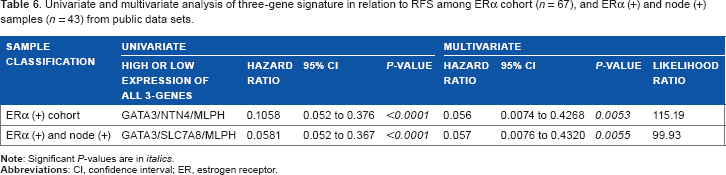

(
Discussion
The current method of determination of ERα status by immunohistochemistry under clinical setup provides information about the expression pattern of ERα with no information on possibly disabled downstream ER pathway.14,17 Thus, it is plausible that the status of the ER pathway is also clinically relevant and may explain variable response to endocrine therapy in ERα (+) patients. Hence, measurements of gene expression profiles that reflect the activity of ER pathway could provide an important insight in understanding the behavior of breast cancers. We reported the expression pattern for seven genes that were regulated by ERα and demonstrated the differential expression in various phenotypes of breast cancer pathology. We observed the highest expression of five genes in ERα (+) and PR (+) cohort of patients (luminal A subtype). Interestingly, among endocrine-treated patients, previous report 25 also showed that the presence of both ERα and PR was a stronger marker for the benefit of adjuvant endocrine therapy than ER alone.
Due to limited availability of clinical information in our data set, we used independent data sets21–23 to assess the predictive ability of our gene signatures, which will give us additional confidence in clinical validity. Meta-analysis of seven genes showed that a gene expression is reliable and robust to determine ER expression in three independent data sets comprising 340 tumor samples. These analyses strengthen our recent findings and suggest that a seven-gene signature can stratify/classify ERα (+) and ERα (–) tumors in an independent data set. Of the seven genes analyzed in the ERα (+) tumors, the high mRNA expression of
Classic parameters, such as ER, PR, HER2 status, number of lymph nodes positive, and tumor size, have been integrated into software applications such as Adjuvant! Online 19 to help doctors in calculating a risk of relapse and benefit from adjuvant therapy. However, uncertainty remains in many cases even with the use of this software. The well-established prognostic and/or therapeutic breast cancer markers are hormone receptors (ER and PR), 26 HER2, 27 Ki-67 antigen, 28 tumor protein p53, 29 carbohydrate 15-3 and carcinoembryonic antigens (CA 15-3 and CEA),30,31 and breast cancer susceptibility genes (BRCA1 and BRCA2). 32 Gene signatures can complement classic prognostic factors to obtain more accurate prognostic information. The 70-gene signature (MammaPrint; Agendia) and the 21-gene signature (OncoType; Genomic Health) are being used in selected patients with early ERα (+) disease to identify those women who will be cured even if they do not receive adjuvant chemotherapy.4,33 These signatures have been extensively studied and are widely used in Europe and USA.34–36 The National Cancer Comprehensive Network guidelines indicate that the 21-gene signature can be considered in women with tumors >0.5 cm, HER2-negative disease, and node-negative disease. 37 Limitations of the 21-gene and 70-gene signatures are intended to be used by women with node-negative breast cancer diagnosis. Many other gene signatures have been developed and have undergone validation. One of them is the breast cancer gene expression ratio test, which only measures the ratio of HOXB13 to IL17BR38,39 A high mRNA expression ratio was associated with a high risk of recurrence in tamoxifen-treated patients. Recently, the accuracy of this test could be improved by including proliferation-associated genes of the molecular grade index, 40 which is an RT-qPCR assay consisting of five genes that are able to identify a subgroup of ER (+) patients with a worse outcome despite endocrine therapy. The Rotterdam 76-gene signature was created on the basis of predicting the development of metastatic disease within 5 years using an unselected patient cohort regarding age, tumor size, grade, and hormone receptor status.41,42
The 5-year survival for patients with the node-negative disease is 82.8% compared with 73% for 1-3 positive nodes, 45.7% for 4-12 positive nodes, and 28.4% for ≥13 positive nodes. 43 These data demonstrate that the risk of recurrence is significant enough with lymph node-positive disease to warrant adjuvant systemic therapy since, generally, a future risk of distant recurrence of 20% or greater is regarded significant enough to consider the risks of therapy. Hence, it is important to stratify ERα (+) and node (+) patients into a low- and high-risk group for RFS. In this study's sample population, out of 109 ER (+) and node (+) samples, 67 samples showed either high or low expression of all GATA3/NTN4/MLPH genes, whereas 43 samples showed either high or low expression of one or two GATA3/SLC7A8/MLPH genes. So the limitation of this gene signature is that not all patient samples will exhibit either low or high gene expression of all three genes and has to be excluded from the prediction.
The
In summary, we show that the high expression of estrogen-responsive three-gene signature
Author Contributions
Conceived and designed the experiments: AT, MP, AB, HR. Analyzed the data: AT. Wrote the first draft of the manuscript: AT. Contributed to the revising manuscript critically for important intellectual content: AT, AB, BM, MP. All authors made an intellectual input and contributed to writing the paper. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgment
Debarshi Chakrabarti provided scientific input, discussions and deliberations during the time of the study.