
Abstract
Head and neck cancers (HNCs) represent a significant and ever-growing burden to the modern society, mainly due to the lack of early diagnostic methods. A significant number of HNCs is often associated with drinking, smoking, chewing beetle nut, and human papilloma virus (HPV) infections. We have analyzed DNA methylation patterns in tumor and normal tissue samples collected from head and neck squamous cell carcinoma (HNSCC) patients who were smokers. We have identified novel methylation sites in the promoter of the mediator complex subunit 15 (MED15/PCQAP) gene (encoing a co-factor important for regulation of transcription initiation for promoters of many genes), hyper methylated specifically in tumor cells. Two clusters of CpG dinucleotides methylated in tumors, but not in normal tissue from the same patients, were identified. These CpG methylation events in saliva samples were further validated in a separate cohort of HNSCC patients (who developed cancer due to smoking or HPV infections) and healthy controls using methylation-specific PCR (MSP). We used saliva as a biological medium because of its non-invasive nature, close proximity to the tumors, easiness and it is an economically viable option for large-scale screening studies. The methylation levels for the two identified CpG clusters were significantly different between the saliva samples collected from healthy controls and HNSCC individuals (Welch's t-test returning P < 0.05 and Mann–Whitney test P < 0.01 for both). The developed MSP assays also provided a good discriminative ability with AUC values of 0.70 (P < 0.01) and 0.63 (P < 0.05). The identified novel CpG methylation sites may serve as potential non-invasive biomarkers for detecting HNSCC.
Introduction
Head and neck cancers (HNCs), in particular, head and neck squamous cell carcinoma (HNSCC) represent a very substantial burden in our modern society, with more than 900,000 cases diagnosed yearly, 1 accompanied by nearly 300,000 deaths. 2 The primary risk factors for the development of HNSCC include tobacco use, alcohol consumption, human papillomavirus (HPV) infections (mainly for oropharyngeal cancers), and Epstein–Barr virus (EBV) infections (for nasopharyngeal cancer). 3 In addition, betel nut chewing, which is common in certain regions of Asia, is also an independent risk factor for the development of HNSCC. 4 The relative prevalence of these risk factors contributes to variations in the observed distribution of HNSCC in different parts of the world. As an example, oral and tongue cancers are common in the Indian subcontinent, nasopharyngeal cancers are common in China and Hong Kong, and pharyngeal and/or laryngeal cancers are prevalent in other populations. 3 One of the major causes for high mortality associated with HNSCCs is the late diagnosis when the tumor has metastasized into other parts of the body.
Human saliva has gained momentum as a diagnostic fluid due to its non-invasiveness, ease of collection, and ability to perform multiple sample collections in large population groups.5–10 In that sense, saliva represents an ideal testing material, especially considering its topical relevance to HNSCCs, often shedding tumor cells into the oral cavity and connected passages. A number of studies described the suitability of saliva for DNA promoter hypermethylation analysis using methylation-specific PCR (MSP), often demonstrating relatively high sensitivity and specificity albeit, utilizing a combinatorial gene screening panels.11,12
In order to provide new diagnostic opportunities for HNSCC, we set out to identify novel CpG methylation sites in the promoter regions of genes selected from known genes that are hypermethylated in a number of cancers, with particular focus on HNSCC. We surveyed a number of publicly available sources, including PubMeth database, and identified 11 candidate genes thought to be hypermethylated in a number of cancers, while no specific information was available on HNSCC. One such candidate gene was MED15/PCQAP, which is situated in the region of 22q11 and is encoding a pleiotropically acting co-factor important for the assembly of the RNA polymerase II complex which is responsible for the expression of all protein-coding genes. 13 Mutations in this gene have also been strongly implicated in the development of lung cancer. 14 Specific pinpointing of the downstream effects of alteration in this gene's function is difficult to achieve due to its involvement in assembly of a core transcriptional machinery of human polymerase II, responsible for transcription of majority of protein-coding genes. To identify clinically meaningful DNA methylation pattern differences associated with HNSCCs, we undertook comprehensive comparative analysis of the methylation in major CpG islands in normal and HNSCC tissue from a number of independently obtained biopsies. Comparison of the methylation patterns in normal and HNSCC cells of the same origin allowed us to account for individual-specific methylation profiles and identify CpG sites whose methylation status correlated exclusively with cancerous cell state (Fig. 1). Our data revealed that the DNA methylation levels for the two identified CpG clusters were significantly different between the saliva samples collected from healthy controls and HNSCC individuals. In addition, the developed MSP assays provided a good discriminative ability, and the identified novel CpG methylation sites may serve as potential non-invasive biomarkers for detecting HNSCC. Identification of novel therapeutic targets and new and specific biomarkers for the early detection of HNC could greatly increase the survival rate and might have a clinical utility as prognostic indicators.

Identification and detection of the differential methylation at the two novel sites in the MED15/PCQAP promoter. (A) Schematic showing location of the sites in the main promoter-associated CpG island. (B) Sequence of the two regions in the MED15/PCQAP promoter illustrating tumor-associated hypermethylation at the two CpG clusters (yellow and green) present in samples of tested subjects (results from three patients', P1–P3, shown). Please note that the reference sequence assumes full methylation at the CG dinucleotides.
Materials and Methods
Study design
This study was approved by the University of Queensland Medical Ethical Institutional Board and by the Princess Alexandra Hospital Ethics Review Board. In the first instance, we collected formalin-fixed, paraffin-embedded (FFPE) tissue sections from HNSCC (n = 6) patients. Certified pathologist assisted in the identification of the tumor and normal sections on the tissue slides. All participants gave informed consent before sample collection. Healthy control subjects (n = 49 for 5′-CpG site and n = 45 for 3′-CpG site) without any clinical signs of cancer as well as HNSCC patients who developed cancer due to smoking (n = 46 for 5′-CpG site and n = 44 for 3′-CpG site) and HPV +ve HNSCC (n = 16) at various clinical stages (stages II–IV) of cancer originating from different anatomical sites within the head and neck regions (oropharynx and oral cavity larynx/pharynx) were recruited in this study.
Saliva sample collection and processing
DN A promoter methylation of PCQAP/MED15 was assayed in DNA isolated from whole mouth saliva (drool) collected from HPV—ve HNSCC patients (both smokers, including recent quitters and non-smokers) as well as HPV +ve HNSCC patients and healthy controls (non-smokers). In general, we have asked volunteers not to eat food prior to donating saliva, adhering to previously published saliva collection protocols from our work.5–10 The volunteers were asked to sit in a comfortable upright position and were asked to rinse their mouths with water to remove food debris. They were asked then to tilt their heads down and to pool saliva in the mouth for about two to five minutes. This, unstimulated saliva samples were collected into Falcon tubes (50 mL, Greiner, Germany) and were transported on dry ice to the laboratory. Samples were then thawed at room temperature and centrifuged at 1500 × g at 4 °C for 10 minutes. The supernatants were discarded, and the cellular pellet was frozen at–80 °C to be later used for MSP analysis.
DNA extraction and bisulfite conversion of saliva samples
DNA extraction and subsequent bisulfite conversions were carried out using the EpiTectPlus kit® (Qiagen GmbH) according to the manufacturer's instructions with the exception of a longer elution incubation time (10 minutes instead of 1 minute) and the use of a slightly larger elution volume (17
Extraction and bisulfite conversion of the DNA from FFPE samples
FFPE tumor and normal samples from HNSCC (n = 6) patients in stages II to IV were retrieved from the Department of Anatomical Pathology at the Princess Alexandra Hospital in Woolloongabba, Australia. FFPE normal tissue sections were also obtained from the same HNSCC patient. A pathologist identified and confirmed the normal part of the tissue from the tumor on hematoxylin and eosin (H&E) stained slides. Tissue sections (5
Identification of the methylation sites in MED15/ PCQAP promoter
In order to identify novel methylation sites, PCR amplification of the main CpG clusters on PCQAP promoter regions was performed on the bisulfite–converted DNA samples from histologically identified tumor and normal tissue using AmpliTaq Gold® 360 Master Mix (Applied Biosys-tems, USA, CAT. 4398901), proofreading polymerase mix. Primer design for bisulfite PCR of the CpG island and subsequent MSP screening were performed using the MethDB (http://www.urogene.org/methprimer/) and BiSearch (http://bisearch.enzim.hu/) online computational resources. Primers listed in Table 1 in supplementary material were used to amplify a ~700 bp region 20,861,600–20,862,400 of human chromosome 22 (GRCh37/hg19, also see Figure 1A and 1B). Using primers listed in Table 1 in Supplementary material, first round of amplification was performed using the following cycling conditions: 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 30 seconds, 60 °C for 2 minutes, 72 °C for 1 minute, followed by a final extension step for 7 minutes at 72 °C using T100™ BioRad thermal cycler. Direct sequencing of the products from six patient biopsy-derived samples was preceded by a second round of amplification adding sequencing-optimized adapter sequences (Table 1 in Supplementary material) and was also performed with identical PCR conditions as first round of amplification followed by sequencing reaction using (Tag) primers. The sequencing reactions were carried out in a 20
MSP analysis of the PCQAP methylation in SCC
Specificity the designed MSP primer pairs was confirmed on the unconverted DNA, which resulted in no gene specific amplifications. Quantification of the MSP product levels was performed using intensity measurements with FUSION-SL chemiluminescence gel imager and Image J 1.47 software (Fiji software). We quantified the methylated, unmethylated, and gDNA loading control PCRs, after running them on 2% agarose gel and stained with GelRed DNA-binding dye. We used “integrated density” value to quantify PCR amplicons. We calculated the ratios of MSP PCR products between methylated to unmethylated for 5′-CpG site and the ratio of methylated to MYOD for 3′-CpG. For both 5′-CpG site and the 3′-CpG sites, PCRs were carried out in a 12.5
Statistical analysis of the results
To assess the significant difference in methylation levels between patients and controls, both unpaired t-test with Welch's correction and non-parametric Mann–Whitney tests were utilized. Difference was considered significant if stringent cut-off of P < 0.05. Data point plots and receiver-operating characteristic (ROC) curves were generated using Graphpad-Prism6 software and online tools (GraphPad, Inc and http://graphpad.com/quickcalcs).
Results
MED15/PCQAP is epigenetically regulated in HNCs
In order to identify genes that are epigenetically regulated in HNCs, we surveyed publicly available DNA methylation databases with a special emphasis on HNSCC.15,16 Amongst other candidates, the MED15/PCQAP gene was found to be methylated in a number of cancers, including HNCs, esophageal, prostate, and buccal cell-derived cancers. It also possesses a clearly identifiable CpG island associated with its main upstream promoter (located at positions chr22:20,861,680–20,862,252 in GRCh37/hg19), comprising 54 CpG dinucleotides. High abundance of the transcription factor and H3K27Ac histone ChIP-seq analyses' hits in that region corroborated our assumption about the key regulatory role of that region in MED15/PCQAP expression.
Identification of HNSCC-specific methylation in MED15/PCQAP promoter
We set out to identify specific methylation patterns within this CpG islands that might be associated with HNSCC tumors by amplifying bisulfte-converted DNA from HNSCC and normal tissues from HNSCC patients. Primers were designed to flank all the CpG sites in the island (see Materials and methods and Table 1 in Supplementary material and Figure 1A and 1B), and used to generate PCR products from both FFPE tumors and normal tissue samples which were then used for the determination of the methylation patterns. Two clusters of two adjacent CpGs each demonstrated consistent tumorspecific methylation, first doubly methylated in five out of six HNSCC patient tumor samples, and second in four out of six HNSCCs (Fig. 1A and 1B). Sequence analysis also revealed several single nucleotide polymorphisms (SNPs) in this region in all six sequenced individual genomes (See Fig. 1A and 1B and Table 1 in Supplementary material).
MSP analysis of methylation at the novel sites within MED15/PCQAP promoter
Upon identification of the differentially methylated CpGs, we proceeded to design MSP strategies allowing us to reliably screen for the presence of both alleles. To achieve this, three primers were designed for each of the CpG doublets. The first common primer was designed within 200 bps from the target CpGs, to work in methylation-insensitive fashion, while the other two primers one for methylated and one for unmethylated versions using MSP algorithms. Specificity of the primers was verified using an ePCR tool for bisulfite-converted DNA PCR prediction on human genome at the BiSearch portal (http://bisearch.enzim.hu/). Efficiency and specificity of the MSPs were validated using a nearly fully artificially CpG methylated HeLa gDNA from New England BioLabs (UK) as positive control and tested also in bisulfte-converted gDNA from a number of “normal” human cells utilized as “negative” controls (pluripotent stem cell line and blood leukocytic fraction) (see Fig. 2). As expected, strong signal was obtained upon amplification of the HeLa DNA with methylated allele-specific MSP primer sets, while the unmethylated set was effective at amplifying negative controls. As anticipated, we obtained a significantly higher methylation in DNA samples from the FFPE tumor as compared to the adjacent normal tissues (see Fig. 1 in Supplementary material).
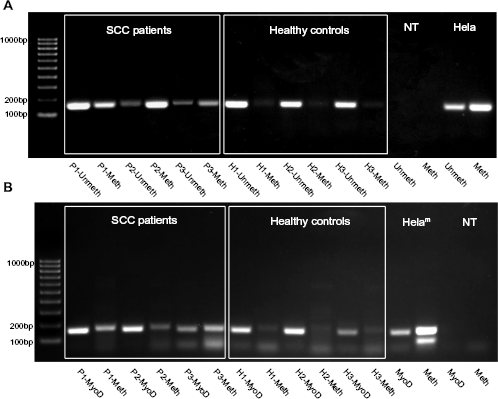
Detection of methylation status of the novel CpG sites using MSP. (
Methylation levels at two novel sites are significantly elevated in saliva of HNSCC patients
Quantitative assessment of the levels of MED15/PCQAP methylation in the DNA from saliva samples of healthy controls and HNSCC patients revealed a statistically significantly higher methylation levels in saliva of HNSCC patients as compared to healthy controls (Fig. 3, the scatter plots of DNA methylation levels at each site). For each of the saliva samples, quantifications of relative methylation levels were performed by comparing the MSPs against methylated and unmethylated forms of the identified novel 5′- and 3′-CpG clusters in MED15/PCQAP promoter (see Materials and methods for further details). Significantly elevated (P < 0.01, Mann–Whitney test) DNA methylation (5′-CpG cluster) was observed in saliva collected from HNSCC patients (HPV –ve) compared with saliva collected from healthy controls. Similarly, significantly elevated (P < 0.05) DNA methylation (3′-CpG cluster) was observed in saliva collected from HNSCC patients (HPV–ve) compared with saliva collected from healthy controls. Significant DNA methylation differences were not observed between saliva collected from HPV +ve patents and healthy controls. Methylation levels in the saliva collected from HNSCC patients (HPV – and +) were higher than in the saliva collected from healthy controls, with means of ~2.7; ~1.9 and ~1.4 for 5′-CpG; and 0.87 and 0.23 and 0.16 for 3′-CpG, respectively (Figure 3A,B). A Kruskall-Wallis test comparing the three groups (controls, HPV –ve and +ve HNSCC patients) resulted in significant (P= 0.00038) median methylation levels.

Detection of the MED15/PCQAP promoter hypermethylation at the novel 5′- and 3′-CpG clusters in saliva from control and HNSCC patient groups. Scatter dot-plot illustrating distribution of the relative methylation levels within control and HNSCC sample groups for (A) 5′ end or upstream and (B) 3′end or downstream. Mann—Whitney test's results shown (**P < 0.001; *P < 0.01; *P= NS).
Predictive power of the new simple MSP-based saliva test
ROC analysis is the most common way of assessing the performance of diagnostic tests. 17 To quantitate the performance of the MSP assay directed specifically to a novel CpG cluster, we generated a standard sensitivity versus specificity plots, which confirmed our notion of high predictive power of the assays (Fig. 4). The areas under the ROC curve parameter appeared to be relatively high at 0.70 and 0.63 for 5′- and 3′-CpGs/MSPs, respectively (Fig. 4), indicating good accuracy of the test in identifying presence of the HNSCC by simple MSP analysis of the DNA from saliva samples. For 5′-CpGs/MSPs, at a cut-off level >1.15, the sensitivity was 70% and specificity was 63%. Similarly, for 3′-CpGs/MSPs, at a cut-off level >0.11, the sensitivity was 68% and specificity was 58%. Based on these data, at 5′-CpGs site appears to be a better biomarker than the 3′-CpG site.

ROC curves for the 5′ (A) and 3′ (B) CpG cluster MSP analysis. AUC – area under the curve value. Both values fall in the fair to good range (0.6–0.7), thus providing an informative predictive power to the testing.
Discussion
MED15/PCQAP gene encodes a member of the mediator complex-comprising family of nuclear proteins having a pleiotropic role in the regulation of RNA polymerase II-mediated transcription. 18 In addition, MED15/PCQAP promoter DNA hypermethylation has specifically been implicated in prostate cancer, endometrial cancer, and various carcinoma aetiologies. 19 It was also found to be one of the targets of Kaposi's sarcoma-inducing herpes viral proteins. 20 We have identified and validated two novel CpG sites (in the 5′ or 3′ end) in the saliva collected from HNSCC patients. Our data revealed that 5′-CpG site is a better diagnostic biomarker compared with the 3′-CpG site. Interestingly, DNA methylation of both of these sites were relatively lower in the saliva collected from HPV +ve HNSCC patients compared with the saliva collected from HPV –ve patients. Important to note is the significant three-way difference between the medians of controls, HPV –ve and HPV +ve patients.
MED15/PCQAP has been strongly implicated in attenuation of at least one of the signaling pathways (TGFβ/Activin signaling) important for initiation and progression of many cancers. 13 Hypermethylation of 5′-CpG site within the promoter of the MED15 gene seems to provide a good correlative readout of the presence of HNSCC, and appears to provide a relatively higher predictive ability than some of the recent reports.21,22 It is particularly interesting because unlike in previous studies,11,21,22 hypermethylation of the MED15 promoter, which is not regulating a typical cancer-suppressor gene, appears to be indicative of a presence of malignant transformation.
HNSCC carcinogenesis is a multistep and multifactorial complex process, encompassing a number of genetic and epigenetic abnormalities in DNA repair, signal transduction, apoptosis, angiogenesis, proliferation, differentiation, and cell cycle regulation. 23 The prognostic assessment of HNSCC is based on the tumor size, anatomic location, and the presence of lymph node metastases. Despite advances in knowledge and treatment of this disease, the five year survival rate for smoking-associated HNSCC remains approximately 50% or less. One reason for the large degree of mortality linked with the smoking-associated HNSCC is late stage diagnosis, which promotes the intrinsic ability of tumor cells to undergo loco-regional invasion. However, current diagnostic strategies rely in part on histological analyses of biopsy samples, which have largely proven inadequate due to the high frequency of patients with recurrent disease. 24
At the present time, there are no early detection methods for HNSCC, and as such, the 5 year mortality rates are high. Therefore, early detection is critical for the improvement of the overall survival of HNSCC cancer patients. As an example, the 5 year survival rate for oral cancer if it is detected at an early stage is 70% compared with 37% for late diagnosis (Data taken from America Cancer Society, updated on 26/2/2013). Saliva is an ideal diagnostic medium to interrogate tumor events as the tumors in the oral cavity are in close proximity to saliva. There is also substantial evidence to justify the use of saliva as a future diagnostic medium for developing diagnostic/prognostic or theranostic tests. As an example, FDA approved OraQuick® HIV test is a testimonial to the success of saliva based assays (http://www.orasure.com). The identified DNA methylation biomarker test can be used either as a point-of-care test, within a routine pathology laboratory, at the GPs office and/or dentist office. Using this DNA methylation assay, if a test result is positive, these individuals can then be referred to specialists for further examinations. Given the increasing rates of HNSCC in the world, the development of a screening program using these DNA methylation biomarkers would have a major international impact on cancer care and cancer control. We envision that a screening program using saliva as a diagnostic fluid would result in a change in current clinical paradigm by introducing for the first time, HNSCC-screening services significantly reducing mortality and morbidity.
Footnotes
Acknowledgments
The authors wish to acknowledge all the staff at the head and neck cancer clinic at the Princess Alexandra Hospital in Woolloongabba and special thanks goes to A/Prof Chris Perry A/Prof Ben Panizza and Ms Dana Middleton (Clinical Trial coordinator). Also to Dr Duncan Lambie for identifying on tissue sections tumor from normal tissues. We also thank Dr Paula Hawthorn and the Australian Equine Genome Research Center at the University of Queensland for providing sequencing facility.
Author Contributions
Analyzed the data: DO, YW, PP. Wrote the first draft of the manuscript: DO. Contributed to the writing of the manuscript: JCW, JGH, CP. Agree with manuscript results and conclusions: CP, WBC. Jointly developed the structure and arguments for the paper: YW, CP, DO. Made critical revisions and approved final version: CP, JGH. All authors reviewed and approved of the final manuscript.
Supplementary Materials
Primers used for bisulphite sequencing and MSP PCRs.

| GENE | NUCLEOTIDE SEQUENCE | PCR PRODUCT SIZE (BP) |
|---|---|---|
|
|
||
| MED15_CpG-Tag |
|
724 |
| 5′-CCA CTC ACT CAC CCA CCC GTA GAA AAT GTA GG-3′ | ||
|
|
||
| 5′-GGG TGG GAG GTG GGA GGG AAC ACA CAA ATA AC-3′ | ||
| Tag sequencing | Forward | |
| 5′- CCA CTC ACT CAC CCA CCC-3′ | ||
| Reverse | ||
| 5′-GGG TGG GAG GTG GGA GGG-3′ | ||
|
|
||
| MED15_MSP5′ |
|
167 |
| 5′-AAA AAT CCC ACA ATC CAA CCC-3′ | ||
|
|
||
| 5′-GTT TTG TGA TTG AGG YGG TGG T-3′ | ||
|
|
||
| 5′-GTT TTG TGA TTG AGG YGG CGG C-3′ | ||
| MED15_MSP3′ |
|
172 |
| 5′-GAT ATG GGT GGT GGG AGT TGG G-3′ | ||
|
|
||
| 5′-AAT CAG ACC CTA ACC TCG CCC G-3′ | 158 | |
| MyoD | Forward | |
| 5′-TGA TTA ATT TAG ATT GGG TTT AGA GAA-3′ | ||
| Reverse | ||
| 5′-CCA ACT CCA AAT CCC CTC TCT AT-3′ | ||