
Abstract
Gallic acid is an organic acid known for its antioxidant and anticancer properties. The present study is focused on evaluating the role of gallic acid in providing better therapeutic outcomes against arsenic-induced toxicity. Animals pre-exposed to arsenic were treated with monoisoamyl
Introduction
Arsenic exposure recently has become a global human health concern of utmost significance. Acute and chronic exposure to arsenic has been associated with various toxic manifestations. An increasing number reports on the toxic effects of arsenic prove its alarming state and the need for relevant therapeutic solutions.1,2 Arsenic-induced oxidative stress forms the major underlying mechanism in most associated adverse manifestations. Arsenic-mediated generation of free radicals gives rise to a variety of reactive oxygen species (ROS) including superoxide (O2•–), singlet oxygen (O2), the peroxyl radical (ROO•), nitric oxide (NO•), hydrogen peroxide (H2O2), dimethylarsenic peroxyl radicals [(CH3)2AsOO•], and the dimethylarsenic radical [(CH3)2As•].2,3
The current management of arsenic poisoning relies on supportive care and chelation therapy.
2
Chelating agents bind to and enhance the urinary and fecal excretion of toxic metals by forming soluble complexes with these metals in vivo.
3
However, no specific chelating agent has been established yet to treat arsenic poisoning. 2,3-Dimercaprol (British anti-Lewisite, BAL) was introduced as an antidote against arsenic-based warfare, but it has serious adverse effects. Other chelating agents such as
Structure of Gallic acid.
Materials and Methodology
Chemicals
Gallic acid, sodium meta-arsenite, and all other chemicals were of analytical grade or of the highest purity available and were purchased from Merck (Germany), BDH Chemicals (Mumbai, India) or Sigma (USA). MiAD-MSA was synthesized under GMP and supplied by Cadila Pharma., Ahmadabad, India. Triple distilled water prepared by Millipore (New Delhi, India) was used throughout the experiments for the preparation of reagents and buffers used for various biochemical assays in our study.
Animals
Adult male Swiss Albino mice weighing 25 ± 5 g drawn from the Defence Research and Development Establishment (DRDE, Gwalior, India) Animal Facility were used in the studies. They were maintained on ad libitum pellet diet (Lipton's India Ltd.) and water in an air-conditioned room with regular alternate cycles of 12 hours of light and darkness. The metal contents of the animal feed (in ppm dry wt) were Cu 10, Mn 55, Co 5, Zn 45, and Fe 70. The animals were weighed every week and the doses adjusted accordingly. All animals received humane care in compliance with the guidelines of the “Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA)”. The Animal Ethics Committee of DRDE approved (RT-03/52/SJSF dated December 17, 2012) the protocols for the experiments.
Experimental design
The animals were divided into two groups and treated for 4 weeks as follows:
Group A: Normal control (n = 24)
Group B: Arsenic as sodium meta-arsenite (1 mg/kg, i.p.) (n = 32)
After 4 weeks, animals from group A and B were divided into the following subgroups and treated daily with gallic acid (95 mg/kg, orally) 10 and/or with MiADMSA (50 mg/kg, orally) 11 for next 10 days:
Group A
Group 1: Control (saline) (n = 8)
Group 2: MiADMSA alone (50 mg/kg, orally for 10 days) (n = 8)
Group 3: Gallic acid (95 mg/kg, orally for 10 days) (n = 8)
Group B
Group 4: Arsenic (saline) (n = 8)
Group 5: Arsenic + MiADMSA (n = 8)
Group 6: Arsenic + gallic Acid (n = 8)
Group 7: Arsenic + gallic Acid + MiADMSA (n = 8)
Twenty-four hours after the last dose, animals from different groups were sacrificed. Blood was collected in non-heparinized and heparinized vials, and tissues were removed, washed, blotted, and stored at –80°C until analysis.
Reactive oxygen species
The amount of reactive oxygen species (ROS) in blood was measured using 2′,7′-dichlorofluorescin diacetate (DCFDA), which gets converted into highly fluorescent DCF by cellular peroxides (including hydrogen peroxide). The assay was performed as described by Socci et al. 12 For the estimation of ROS in blood, 5% red blood cell (RBC) hemolysate was prepared and diluted to 1.5% with ice-cold 40 mM Tris-HCl buffer (pH 7.4). For tissue ROS estimation, 10% tissue homogenate was prepared. The tissue was homogenized (10 mg) in 1 mL of ice-cold 40 mM tris–HCl buffer (pH 7.4), further diluted to 0.25% with the same buffer and placed on ice. Then, 40 µL of 1.25 mM DCF-DA in methanol was added for ROS estimation. All samples were incubated for 15 minutes in a 37°C water bath. Fluorescence was determined at 488 nm excitation and 525 nm emission wavelengths using a fluorescence plate reader (Perkin Elmer, LS-55, UK).
Blood glutathione
Analysis of blood glutathione (GSH) concentration was performed by slightly modifying the method of Jollow et al.
13
In brief, 0.2 mL of whole blood was added to 1.8 mL of distilled water and incubated for 10 minutes at 37°C for complete hemolysis. After hemolysis, 3 mL of sulfosalicyclic acid was added and the mixture was centrifuged at 2500
Blood δ-aminolevulinic acid dehydratase
The activity of δ-aminolevulinic acid dehydratase (δ-ALAD was determined by the method of Berlin and Schaller. 14 The assay system consisted of 0.2 mL heparinized blood and 1.3 mL distilled water. The mixture was incubated at 37°C for 10 minutes for complete hemolysis. After 10 minutes, 1.0 mL of standard ALA solution was added to the experimental tubes and 1.0 mL trichloro acetic acid (TCA) to blank tubes. The mixture was incubated again for 60 minutes at 37°C. The reaction was stopped after 60 minutes by adding 1.0 mL of TCA in experimental tubes and 1.0 mL of ALA in the blank. The mixture was centrifuged, and a 1.0 mL aliquot was taken in a test tube and an equal volume of Ehrlich reagent was added to it. After 5 minutes, the absorbance was read at 555 nm.
Thiobarbituric acid reactive substance
Measurement of lipid peroxidation was carried out by the method of Ohkawa et al 15 in blood and tissue. One milliliter of the tissue homogenate, prepared in 0.15 M KCl (5% w/v), was incubated for 1 hour at 37°C followed by the addition of 10% TCA, which was mixed thoroughly and centrifuged at 3000 rpm for 10 minute. One milliliter of thiobarbituric acid (TBA) was added to 1 mL of the supernatant, and the mixture was kept in a boiling water bath for 10 minutes till a pink color appeared. One milliliter of double-distilled water was added to the mixture after cooling the tubes. Malondialdehyde (MDA) formation was determined by reading the absorbance at 535 nm. The amount of thiobarbituric acid reactive substance (TBARS) was calculated using a molar extinction coefficient of 1.56 x 10 5 /M/cm. The absorbance of the supernatant was read at 535 nm, and the values were expressed as nanomols of MDA/mL blood or µg/gm tissue wt in tissue samples.
Tissue glutathione
Tissue GSH content was measured as described by Hissin and Hilf.
16
Briefly, 0.25 g of tissue sample was homogenized on ice with 3.75 mL of phosphate–EDTA buffer and 1 mL of 25% HPO3, which was used as a protein precipitant. The total homogenate was of H2O2 (1 mM) and 0.3 mL of tissue supernatant. After incubation at 37°C for 15 minutes, the reaction was terminated by the addition of 0.5 mL of 5% TCA. Tubes were centrifuged at 1500
Glutathione peroxidase and glutathione S-transferase
Glutathione peroxidase (GPx) was determined by the method of Flohe and Gunzler
17
at 37°C. The supernatant that was obtained after centrifuging 5% tissue homogenate for 10 minutes at 1500 g, followed by 10,000
Glutathione S-transferase (GST) activity was determined by a protocol described by Habig et al.
18
The supernatant was obtained after centrifuging 5% tissue homogenate at 1500
Superoxide dismutase (SOD)
Brain and kidney SOD activity was assayed by the method of Kakkar et al.
19
Briefly, the supernatant was obtained after centrifugation (1500
Catalase
Catalase (CAT) activity in brain and kidney was assayed following the procedure of Aebi. 20 A reaction mixture containing 1 mL of phosphate buffer, 0.1 mL of hemolysate and tissue homogenate, 0.4 mL of distilled water, and 0.2 mL of H2O2 was prepared. A control mixture was prepared containing 1 mL of phosphate buffer, 0.5 mL of distilled water, and 0.2 mL of H2O2. Mixtures were further incubated at 37°C for 15 minutes, and the reaction was stopped by the addition of 2 mL of acetic acid with dichromate (1:3 ratio of 5% potassium dichromate in distilled water and glacial acetic acid). The above mixture was boiled for 15 minutes and then cooled. The amount of H2O2 consumed was determined by recording the absorbance at 570 nm.
Total protein
Total protein was measured by the method of Lowry et al. 21 Briefly, 0.2 mL of the tissue homogenate was added to an equal amount of cold TCA and kept for a minimum of 2 hours or overnight, which resulted in the precipitation of the protein. The mixture was centrifuged at 2,000 rpm for 10 minutes, and the obtained pellet was dissolved in NaOH. A standard protein solution was prepared using bovine serum albumin (BSA). Different aliquots of the standard protein solution were taken, finally the volume was made up to 1.0 mL with distilled water, and 5.0 mL of alkaline Lowry's reagent was added. After shaking, the mixture was incubated for 10 minutes at room temperature. After adding 0.5 mL of Folin-Ciocalteu reagent (appropriately diluted) and thorough mixing, the reaction mixture was incubated for 30 minutes at room temperature in the dark. The intensity of color depends on the amount of aromatic amino acids present. Absorbance was measured at 650 nm.
Urinary8-oxo-7,8-dihydro-2deoxyguanosine(8-OHdG)
It was measured using the 8-OHdG ELISA kit (Merck).
Serum glutamic oxaloacetic transaminase (SGOT), serum glutamic pyruvic transaminase (SGPT), and lactate dehydrogenase (LDH)
GOT, GPT, and LDH activities were measured in serum using Merck kits.
Estimation of metals
Arsenic concentrations in blood, liver, brain, and kidneys were measured after wet acid digestion using a microwave digestion system (CEM, model MDS-2100). Samples were brought to a constant volume, and the determination of blood and tissue arsenic was performed using an autosampler (AS-72) and a graphite furnace (MH) fitted with an atomic absorption spectrophotometer (AAS, Perkin Elmer model AAnalyst 100).
Statistical analysis
Experimental results are expressed as the mean ± SEM and are accompanied by a number of observations. Data was assessed by the method of one-way analysis of variance (ANOVA) using the Graph pad Instat software. On significant difference among the group mean, the unexposed and exposed group (with or without treatment) were compared by Dunnett's multiple comparison test. Values with matching symbol notation in each column were not significant at the 5% level of probability.
Results
Arsenic exposure resulted in changes in the biochemical endpoints in blood, suggesting oxidative stress (Fig. 2). ROS and GSH levels increased, while ALAD activity showed significant depletion on arsenic exposure. Oxidative stress reflects a disturbance in the balance between the systemic production of ROS and antioxidant defenses against these free radicals. The assessment of GSH levels is a useful indication of the redox potential and the cell's ability to prevent oxidative stress. Inhibition of ALAD enzyme by arsenic leads to decreased heme synthesis and ultimately anemia. ALAD inactivation may also lead to the accumulation of ALA, which can cause an overproduction of ROS, which in part could explain arsenic-induced oxidative stress. While the combined administration of MiADMSA and gallic acid led to an increased ROS level, GSH too showed an increase. Administration of MiADMSA, gallic acid, and their combination proved effective in increasing ALAD activity toward normal level.
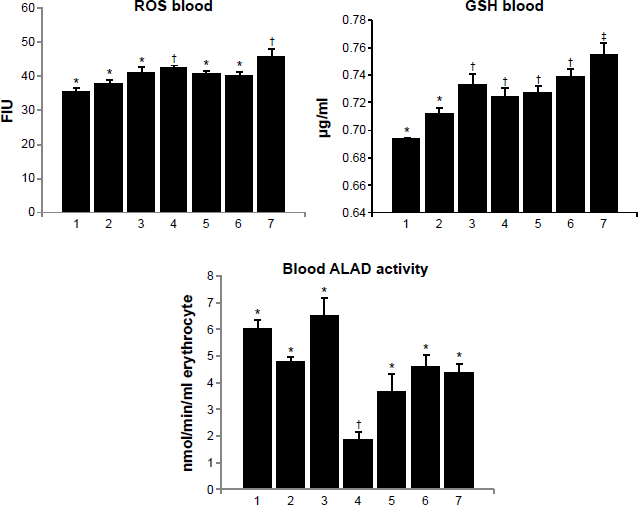
Effect of MiADMSA alone or in combination with gallic acid on blood ROS, GSH, and ALAD levels in arsenic-exposed mice. Groups: 1, control (no treatment); 2, MiADMSA; 3, gallic acid; 4, arsenic; 5, arsenic + MiADMSA; 6, arsenic + gallic acid; 7, arsenic + gallic acid + MiADMSA.
The data in Table 1 suggest pronounced liver injury on arsenic exposure based on increased serum LDH, GOT, and GPT activities accompanied by an increased hepatic arsenic concentration (Table 2). Aspartate transaminase (AST) or alanine transaminase (ALT) activity is a valuable aid primarily in the diagnosis of liver disease. Although not specific for liver disease, they can be used in combination with other enzymes to monitor the course of various liver disorders. While all three treatments (MiADMSA and gallic acid alone and gallic acid + MiADMSA in combination) were able to reduce these alterations significantly, no single treatment was more effective than the other two in eliciting these effects more efficiently. Blood arsenic concentrations showed significant depletion in arsenic-exposed groups following treatment with MiAD-MSA, both individually and in combination with gallic acid (Table 2).
Effect of MiADMSA alone or in combination with gallic acid on serum biochemical variables (LDH, GOT, and GPT) and arsenic concentration in arsenic-exposed mice.
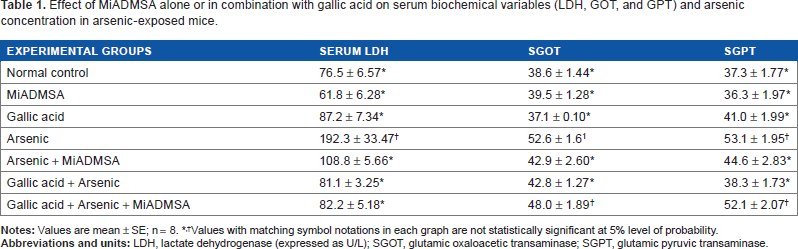
Values with matching symbol notations in each graph are not statistically significant at 5% level of probability.
Effect of MiADMSA alone or in combination with gallic acid on arsenic concentration in blood, liver, kidney, and brain in arsenic-exposed rats.
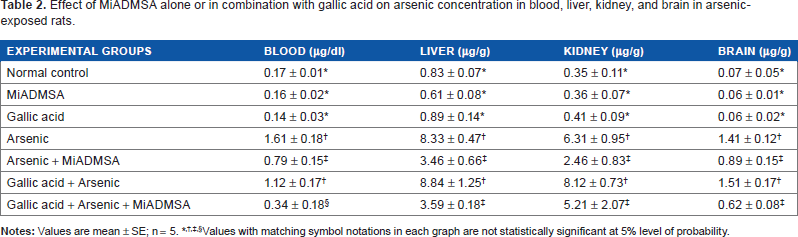
Values with matching symbol notations in each graph are not statistically significant at 5% level of probability.
Brain ROS level increased on arsenic exposure. It was, however, interesting to note that the administration of gallic acid in normal animals also led to an increase in ROS level. This might be due to fact that we used a higher dose of gallic acid. It will be interesting to see whether such an effect persists at lower doses. Brain GSH level decreased significantly on arsenic exposure (Fig. 3). No effect of any of the treatment on the brain TBARS level was noted where the end product of lipid peroxidation is MDA. Treatment with combined administration of gallic acid and MiADMSA produced a significant recovery in brain GSH level, while no effect on brain ROS level was noted.
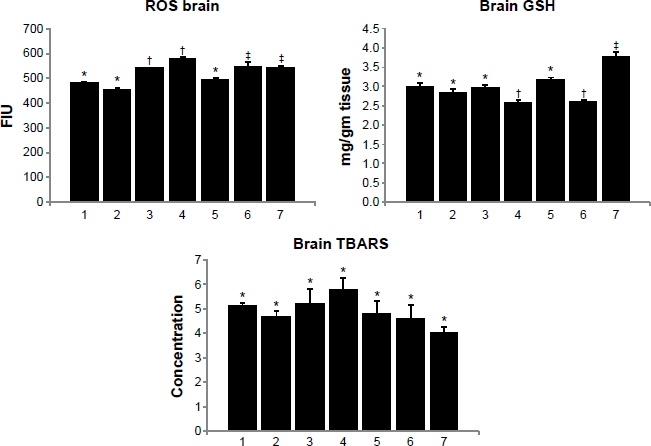
Effect of MiADMSA alone or in combination with gallic acid on brain ROS, GSH, and TBARS levels in arsenic-exposed mice. Groups: 1, control (no treatment); 2, MiADMSA; 3, gallic acid; 4, arsenic; 5, arsenic + MiADMSA; 6, arsenic + gallic acid; 7, arsenic + gallic acid + MiADMSA.
Liver ROS increased significantly on arsenic exposure, and it remained unchanged on treatment with gallic acid and MiADMSA either alone or in combination. Liver GSH, on the other hand, increased on arsenic exposure but responded favorably to monotherapy with MiADMSA (Fig. 4). Interestingly, liver TBARS, which increased on arsenic exposure, showed further elevation on combined treatment with MiADMSA and gallic acid.
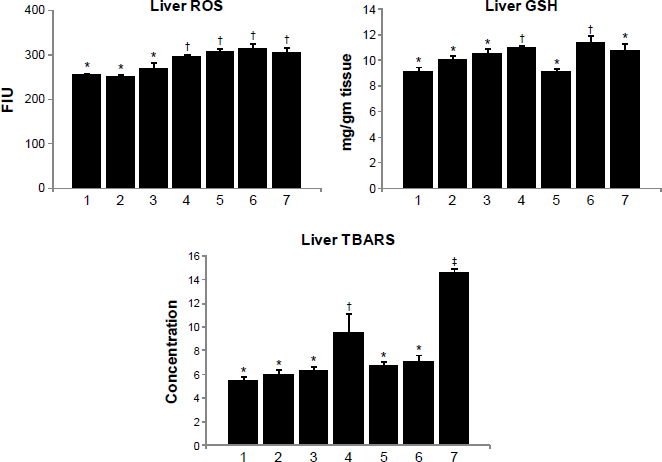
Effect of MiADMSA alone or in combination with gallic acid on liver ROS, GSH, and TBARS levels in arsenic exposed mice. Groups: 1, control (no treatment); 2, MiADMSA; 3, gallic acid; 4, arsenic; 5, arsenic + MiADMSA; 6, arsenic + gallic acid; 7, arsenic + gallic acid + MiADMSA.
Table 3 shows the effects of arsenic exposure and different treatments on the activities of different antioxidant enzymes in mouse brain and liver. Brain SOD, CAT, and GPx activities showed significant inhibition, while GST activity increased significantly on arsenic exposure. The activity of brain SOD, a class of closely related enzymes that catalyze the breakdown of the superoxide anion into oxygen and hydrogen peroxide, showed further inhibition on gallic acid and MiADMSA + gallic acid administration. On the other hand, a marked improvement in CAT, whose functions include catalyzing the decomposition of hydrogen peroxide to water and oxygen, GPx (which catalyzes the breakdown of hydrogen peroxide and organic hydroperoxides), and GST activities was noted post MiADMSA and/or Gallic acid treatment. Administration of gallic acid had little or no effect on these variables.
Effect of MiADMSA alone or in combination with gallic acid on brain and liver antioxidant enzyme (SOD, CAT, GPx and GST) activity in arsenic exposed mice.
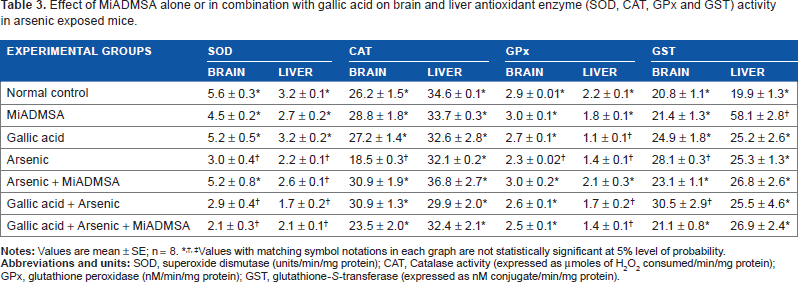
Values with matching symbol notations in each graph are not statistically significant at 5% level of probability.
Liver SOD and GPx activities decreased, while GST increased on arsenic exposure. There was no effect of any of the treatments on hepatic SOD activity. Treatment with MiADMSA alone was able to restore GPx activity toward normal level (Table 3). Liver GST activity, which showed marginal increase on arsenic exposure, remained unchanged on treatments.
8-OHdG is an oxidized derivative of deoxyguanosine. It is one of the major products of DNA oxidation. The concentration of 8-OHdG within a cell is used as a measure of oxidative stress. Its concentration in DNA is, in fact, a quantitative analysis of the degree of DNA damage that an organism has undergone. We measured its level in urine. A significant increase in the 8-OHdG level was noted when arsenic and gallic acid were administered alone (Fig. 5). The level was significantly reduced on monotherapy with MiADMSA in arsenic pre-exposed animals, while gallic acid administration alone and in combination with MiADMSA did not have any effect on the elevated 8-OHdG level.
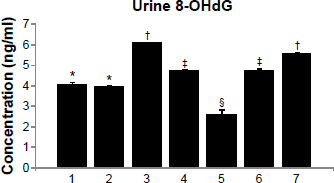
Effect of MiADMSA alone or in combination with gallic acid on urinary levels of 8-OHdG in arsenic exposed mice. Groups: 1, control (no treatment); 2, MiADMSA; 3, gallic acid; 4, arsenic; 5, arsenic + MiADMSA; 6, arsenic + gallic acid; 7, arsenic + gallic acid + MiADMSA.
Table 2 shows the effect of gallic acid and MiADMSA on blood and tissue arsenic concentrations. Significant increase in blood, brain, kidney, and liver arsenic concentration was observed in arsenic-exposed animals. A significant depletion of arsenic was noted on MiADMSA administration in the above organs. Gallic acid, on the other hand, proved ineffective in depleting arsenic from these organs either on monotherapy or in combination with MiADMSA.
Discussion
Arsenic is an element found in the earth's crust and biosphere, and has been known as a human poison for centuries. 22 An abundance of arsenic results in the possibility of daily exposures to humans, which may be via ingestion through drinking water (major route) or through inhalation and skin absorption (minor route). 23 The present study, for the first time, evaluated the effects of gallic acid in arsenic-induced oxidative stress. Gallic acid was screened for its efficacy in two treatment regimens, ie, as monotherapy (administered alone) and in combination with a chelating agent MiADMSA, against arsenic-induced oxidative stress in mice.
The central nervous system and liver have been the major target sites for arsenic to exert its toxicological effects. 24 Arsenic exposure produces a large amount of ROS and reactive nitrogen species, which can impair cellular antioxidant defense systems and simultaneously damage the cellular components such as lipids, proteins, and DNA. 25 In the present study, we observed that arsenic administered orally to mice resulted in high ROS and subsequent damage, indicative of oxidative stress. Results from the present study did not clearly suggest any beneficial effect of gallic acid in arsenic poisoning when tested as monotherapy or in combination with MiADMSA. MiADMSA, however, demonstrated a clear benefit and showed reliable efficacy in reversing arsenic-induced oxidative stress biomarkers in blood, brain, and the liver. Discussing the results more in detail, however, gives us some interesting inferences from the study observations.
Arsenic is known to adversely affect the hematopoietic system by inhibiting various enzymes involved in its physiological pathways. ALAD is one such enzyme that has been established as a biomarker for lead and arsenic hematopoietic toxicity. In the present study, exposure to arsenic at the dose of 10 mg/kg (orally) repeatedly for 30 days caused significant inhibition of blood ALAD activity. This enzyme plays a crucial role in the heme biosynthesis pathway. Arsenic is known to inhibit blood ALAD activity by virtue of its free thiol binding affinity, thus also targeting other thiol-containing functional proteins or biomolecules (Fig. 2).26,27 The effect on blood and tissue GSH observed in the present study further supports the argument. 28 Inhibition of ALAD activity leads to the accumulation of ALA, which may be an added contributing factor for arsenic-induced oxidative stress. Monteiro et al 29 first reported that the conversion of oxyhemoglobin to methemoglobin is mediated by ALA autoxidation, producing ROS. Additionally, arsenic-induced oxidative stress is known to be mediated via ROS.30,31 Arsenic produces ROS directly during the methylation process or as metabolic intermediates, or indirectly by depleting GSH and other antioxidant enzymes. In the present study, arsenic-induced inhibition in blood ALAD activity was significantly recovered by all the treatment groups. MiADMSA monotherapy has been previously reported to effectively counter arsenic-induced stress. However, this is the first report that demonstrates the beneficial effect of gallic acid as protection against arsenic-induced ALAD inhibition. This could be due to the antioxidant property of gallic acid. MiADMSA, on the other hand, may be effective by virtue of the thiol groups in its structure and also via arsenic depletion from the biological site, facilitating the synthesis and renewal of the enzyme in the system.
Biochemical assays in blood suggested arsenic-induced oxidative stress, which may be seen as toxicity to blood as a tissue or as representative of arsenic-induced systemic toxicity. Arsenic elicited high ROS generation in blood compared to normal control. Arsenic-induced ROS generation, as discussed, may be the direct effect of arsenic metabolites that function as free radicals or indirectly via oxygen radical generation and antioxidant depletion. 2 This high amount of ROS generated in turn elicits antioxidant defense mechanisms such as increase in antioxidant production such as GSH, as seen in the present study. Also, MiADMSA and gallic acid monotherapy significantly reduced the arsenic-induced ROS generation, but failed to normalize blood GSH levels. On the other hand, combination therapy with gallic acid and MiAD-MSA did not counter arsenic-induced ROS and GSH in the blood. It may be suggested that owing to the –SH and –OH group, respectively, MiADMSA and gallic acid targeted the first step of free-radical neutralization, whereas the second line effect of restoring the pro- to antioxidant balance could not be achieved by either therapeutic agent evaluated against arsenic. In combination, their inefficacy may either represent an adverse pharmacological interaction or simply the effect of a high dose of gallic acid, which in presence of MiADMSA produced stress to the animal.
Tissue oxidative stress was evaluated in brain and liver since both are the most vulnerable targets to arsenic toxicity. Brain has relatively poor antioxidant defense 32 and contains large amounts of polyunsaturated fatty acids and consumes 20% of the body's oxygen. 33 Liver, on the other hand, being the prime site for the metabolism of arsenic, has the closest interaction with various arsenic species. During arsenic metabolism in the liver, conversion of As 3 + to As 5 + under physiological conditions leads to ROS generation. In the present study, arsenic intoxication induced high ROS generation, as seen in both the brain and liver of mice. Interestingly, arsenic-induced ROS was counteracted effectively by MiADMSA followed by gallic acid mono and combination therapy; however, high ROS due to arsenic in the liver was not reduced by any of the treatment regimens. This may be explained as due to the close proximity of hepatocytes to arsenic species, causing extensive damage. The brain barrier, on the other hand, only allows limited amount of arsenic and takes longer exposure times to reach the target site. Further, the fact that the combination treatment was less effective than MiADMSA can be attributed to the high ROS observed in mice brain exposed to gallic acid alone. This suggests that gallic acid may induce some brain toxicity on its own at the given dose or regimen. Thus, despite its antioxidant nature, its property 34 as an anticancer (cytotoxic) compound may play a certain role. However, interestingly, gallic acid monotherapy also reduced arsenic-induced brain ROS.
Arsenic induced a significant decrease in the glutathione levels in brain (Fig. 3), whereas an increase in liver GSH was observed in arsenic-exposed mice (Fig. 4). This may be attributed to the limited stores and capacity of the brain antioxidant defense compared to that of other robust tissue such as liver that is involved in metabolism and detoxification of xenobiotics. Monotherapy with MiADMSA significantly reversed the arsenic-induced toxic effect on tissue GSH. Gallic acid, however, was effective only in combination at the hepatic site but was ineffective in both therapeutic regimens (alone and in combination) in the brain. Other antioxidants such as GPx, SOD, and CAT were also adversely affected by arsenic toxicity, which decreased their activity by binding to thiol group. 35 Significant depletion of these antioxidants was observed in both brain and the liver. Antioxidant enzymes are considered to be the first line of cellular defense against oxidative damage. SOD is an antioxidant metalloenzyme that reduces superoxide radicals to water and molecular oxygen. 36 CAT is a hemoprotein that reduces hydrogen peroxide to molecular oxygen and water. 37 The reduction in SOD activity in brain tissue of arsenic-exposed animals may be due to the enhanced production of superoxide radical anions. 38 Arsenic intoxication also significantly reduced the CAT activity in the brains of experimental rats. GST and GPx are also antioxidant enzymes that counteract free-radical generation. GST and GPx play major roles in the reduction of organic hydroperoxides within membranes and lipoproteins in the presence of GSH. 39 A significant inhibition of these enzymes results in the accumulation of ROS. Elevated ROS levels may cause peroxidation of the lipid molecule, which is an important structural component of the cell membrane.2,28 In the present study, arsenic-exposed animals showed significant depletion of these enzymes, which was recovered only partially by the therapeutic regimens (Table 3). A better recovery was observed in brain antioxidant levels compared to liver where partial or no improvement was observed. MiADMSA was found to be most effective in restoring the brain antioxidant enzymes followed by the gallic acid and combination groups, which were found effective possibility due to the intrinsic antioxidant properties of gallic acid. Liver therefore is indicated to be damaged to a greater extent compared to brain in the present study.
TBARS is suggested to be a marker of lipid peroxidation that follows the ROS generation and its subsequent effect post-antioxidant defense failure. Increase in TBARS levels post arsenic exposure as seen in liver suggests membrane damage due to ROS generation. This can further be supported by a moderate increase in serum AST and ALT activities, which are important liver enzymes secreted and released into serum in case of hepatocyte injury. MiADMSA and gallic acid monotherapy post arsenic exposure significantly reduced the elevated TBARS levels, suggesting recovery from stress. However, combination treatment resulted in higher TBARS levels, thus indicating elevated toxicity. Serum AST and ALT levels show similar trends when compared with TBARS levels (Table 1 and Fig. 4).
As shown in Table 1, LDH, which is an indicator of cell necrosis, was found significantly elevated in arsenic-exposed animals. LDH is an enzyme that catalyzes the interconversion of pyruvate and lactate with concomitant interconversion of NADH and NAD+; medically it is used as a biomarker of tissue injury. It is present in most tissue cells including RBCs, and thus a rise in serum LDH may suggest hemolysis as well as other tissue damage. This is unlike GOT and GPT, which are specific markers to hepatotoxicity. In the present study, arsenic-induced increase in LDH was significantly decreased by all the treatment groups but most effectively by the combination groups followed by gallic acid and MiADMSA monotherapy, suggesting protection.
Overall, the present study clearly suggests arsenic-induced blood and tissue oxidative stress, as supported by numerous previous reports. The protective and therapeutic potential of gallic acid as evaluated in mono and combination therapy regimens shows assorted results. However, inferring from them, we may suggest that gallic acid shows partial protection from arsenic intoxication in mice along with signs of mild toxicity, as seen in brain ROS and liver TBARS. The protection observed may be attributed, as previously shown, to the antioxidant properties of gallic acid due to its capacity to scavenge ROS as superoxide anions, hydrogen peroxide, hydroxyl radicals, and hypochlorous acid.40,41 Gallic acid has tri-hydroxyl groups, which are suggested to be phenolic hydroxyl groups having a radical scavenging effect. 42 The hydroxyl group at the para position to the carboxylic group is especially effective for the antioxidant activity of gallic acid.43,44 On the other hand, the signs of mild stress, especially when administered in combination with MiADMSA, may be credited to its pro-oxidant and anticancer properties, which render it cytotoxic.44–46 Further, we speculate that gallic acid may provide better protection without adverse effects at possibly lower doses, which must be investigated in the future studies.
Conclusion
Gallic acid through its antioxidant and anticancer property may be a promising candidate for therapy against arsenic. In the present study, however, we observed little or no additional beneficial effect of gallic acid when given in combination with MiADMSA in reducing arsenic burden and reducing the toxic effects of arsenic. We, however, still recommend the use of gallic acid during MiADMSA monotherapy in view of its safety profile and antioxidant effects.
Author Contributions
Conceived and designed the experiments: SJSF. Analyzed the data: SJSF and VP. Wrote the first draft of the manuscript: VP. Contributed to the writing of the manuscript: SJSF. Agree with manuscript results and conclusions: SJSF. Jointly developed the structure and arguments for the paper: SJSF. Made critical revisions and approved final version: SJSF. Both authors reviewed and approved of the final manuscript.