
Abstract
RNA northern blots provide robust measurements of gene expression. The simple northern blot technique described in this report has been optimised to provide rapid, reproducible detection and analysis of mature and precursor forms of microRNAs. This protocol economises on the use of commercially available components and secondly reduces the hybridisation step to 2 hours.
Keywords
MicroRNAs are small non-coding RNA genes which play a profound role in gene regulation.1,2 The detection of miRNA genes is an important step in the understanding of their role and function. A well-established and robust method to measure gene expression is by Northern blot. This method is one of the gold standards for elucidating the presence and level of miRNA genes. In this report, we present a simple and rapid northern blot protocol using the XCell SureLock™ Mini-Cell apparatus which has been modified for the detection of miRNA genes in mammalian cell lines.
In brief, total RNA is separated using a precast 15% TBE Urea gel (Invitrogen, U.S.A) with the XCell SureLock™ Mini-Cell apparatus (Invitrogen, U.S.A). After electrophoresis RNA is transferred onto a nylon membrane using the same unit. A reverse complement probe to the miRNA mature sequence is end-labelled and incubated with the membrane at 37 °C for 2 hours.
The advantage of this approach is that it utilises one electrophoretic unit for both RNA electrophoresis and transfer. As the electrophoretic unit is a compact system, it further reduces the volume of reagents required for this procedure. There is no requirement for enrichment of the small RNA species and this technique also permits the detection of precursor miRNAs which are typically 65–70 nt in length.
Total RNA isolated from cell lines using Trizol (Invitrogen, U.S.A.) was quantitated at 260 nm using a standard spectrometry. Typically, 10 μg of total RNA prepared in 2 X TBE urea sample buffer (Invitrogen, U.S.A.) to a final volume of 10 μl. Samples were heated at 70 °C for 5 minutes and loaded onto a precast 15% (w/v) polyacrylamide gel containing TBE-urea (Invitrogen, U.S.A). The application of a high percentage gel is critical for the separation of both the precursor and mature miRNA sequences.
Prior to loading of samples, wells were repeated flushed with 1 X TBE Running Buffer (Invitrogen, U.S.A). Electrophoresis was performed in 1 X TBE Running Buffer using the XCell II™ Mini-Cell apparatus (Invitrogen, U.S.A). The voltage was set at 180 V with a run time between 60–90 min. After electrophoresis, the gel was rinsed in deionised water, followed by a 5 minute wash in 1 X TBE. RNA was transferred onto a nylon membrane using the XCell II™ Mini-Cell apparatus (Invitrogen, U.S.A) for 1.5 hours at 20 V. The nylon membrane was placed into the Stratagene UV Cross linker® and RNA cross-linked at 1200 kJ. Alternatively, the membrane was baked at 80 °C for four hours. The membrane can be probed immediately or stored at −20 °C until needed.
For design of the miRNA probe, mature miRNA sequences were retrieved from miRBASE 3 and a reverse complement function performed. For example, miR-205 mature sequence is 5′ UCCUUCAU-UCCACCGGAGUCUG 3′. Thus the probe sequence is 5′ CAGACTCCGGTGGAATGAAGGA 3′. All probes were composed of standard DNA bases with no modifications. For radiolabelling of short oligonucteotides the preferred method was end labelling using the forward reaction with T4 polynucleotide kinase (PNK). In detail, 100 ng of oligonucleotide was added to a reaction mix containing 1 μL of PNK (10 U) (New England Biolabs, U.S.A), 2 μL of 10X PNK buffer (New England Biolabs, U.S.A), 4 μL of 32P dATP (10 mCi/ml), to a final volume of 20 μL. The mix was incubated at 37 °C for 1 hour. The reaction was inactivated by incubating at 65 °C for 10 minutes. The labelled miRNA probed was filtered through a NICK™ column (Pharmacia Biotech, U.S.A) to remove unincorporated nucleotides. Typically, an elution profile was generated with the majority of the labelled probe eluted in the second or third fraction. The collection of these fractions is crucial as it provides a high degree of purity for the labelled probe. These fractions were combined and utilised in the hybridisation stage.
A prehybrisation step was performed by incubating the membrane with 10 mL of ExpressHyb solution (Clontech, U.S.A.) pre-heated to 65 °C. Prehybridisation was performed for at least 1 hour at 65 °C in a standard rotating hybridisation oven. The probe was added directly to the prehybridisation solution and the membrane hybridised for at least 2 hours at 37 °C with rotation in a hybridization oven. After hybridisation the solution was discarded and the membrane transferred to a wash container. The membrane was washed with 500 ml 2 X SSC/0.05% SDS for 5–10 minutes at room temperature. Radioactivity was measure directly from the membrane using a Geiger counter. If the membrane emitted low to moderate activity the membrane was covered in a plastic wrap and exposed to a phosphorimaging screen (Amersham Biosciences, Sweden). However, if a high level or excess activity was observed, the membrane was subsequently washed with 500 ml 2 X SSC/0.05% SDS for 5 minutes at room temperature. In our experience, two washes are sufficient to remove nonspecific background activity. The membrane was exposed from 1–2 days prior to image acquisition on the phosphoimager (Fig. 1a).
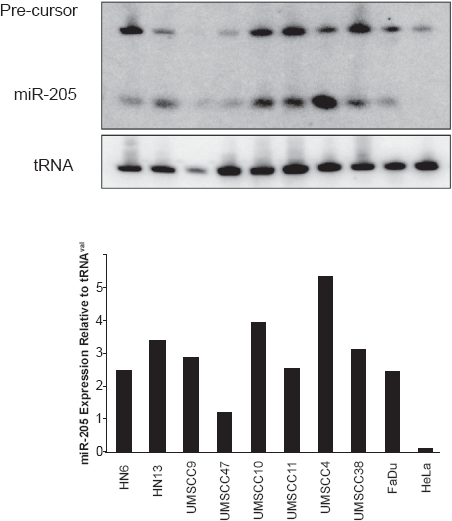
Fast and simple northern blots for miRNA detection. A) Detection of mature miR-205 and its precursor species. B) RNA loading control using tRNAval. C) Quantitation of the expression levels using ImageQuant.
MicroRNA radiolabelled probes were removed from membranes by washing in a high percent detergent solution. The membrane was soaked for 1 hour in 500 mL 0.5% SDS (pre-heated to 100 °C) on an orbital shaker. Excess liquid was drained and the membrane covered in plastic wrap. The membrane was exposed to a phosphorimaging screen overnight to verify if probe removal was successful.
RNA loading was then adjusted using tRNAval (5′CTAAGTGTAAGTTGGGTGCTTTGTGT-TAAGCTACACTCTG 3′). This probe was end labelled using the PNK method and hybridised to the membrane as described previously (Fig. 1). In general, tRNAval probe has a high level of activity after the washes and exposure time is approximately 12 hours or overnight. Interestingly we observed that expression of tRNAval was fairly consistent across all the cell lines tested in this study (Fig. 1b).
Figure 1 is a representative northern blot result using this scale down approach. Detection of both the mature miRNA and precursor species is shown. Depending on the miRNA, the presence of the precursor structure is not observed. These expression profiles can also be quantitated using software such as ImageQuant to provide a graphical representation of the data (Fig. 1c). We have now employed this method to successfully detect 18 miRNA species in 16 different human cell lines and the S2 drosophila line. 4 From our experience, miRNAs which are upregulated are easier to detect on Northern blots. In Figure 1, there is a four fold difference in the sensitivity of miR-205.
To increase the sensitivity and specificity, several studies have now utilised LNA (locked nucleic acid)-modified oligonucleotides.5,6 This has been shown to increase both the specificity and sensitivity of miRNA hybridisation. Detection of specific miRNAs were achieved when using 2.5 μg of total RNA. This may be advantageous if samples are derived from patients where the RNA quantity may be in limiting quantities.
There are several technical advantages in using this classical Northern blot approach to measure gene expression. Firstly, the membrane can be stripped and reprobed for the expression of a different miRNA gene. From our experience, the membrane can be utilised 3 times before the quality of the RNA is compromised. Moreover, total hybridisation time is only 2 hours and can be reduce to 1 hour if the pre-hybridisation step is excluded.
Secondly, the use of precast gels reduces reagent preparation time and provides consistency in the small RNA separation. In addition, due to the small size of the transfer unit, there is a significant reduction in the volume of reagents needed.
Lastly, northern blot detection for examining single gene miRNA expression and validating array results is a well-established and reliable method. In addition, to this benchmark report, an earlier report also demonstrated the utility for Northern blots in the detection of HIV-1-encoded miRNA genes. 7
In summary, this simple and rapid, northern blot technique will provide consistence measurement for the expression of miRNA genes.
Footnotes
Disclosure
The author reports no conflicts of interest.