
Abstract
Signal transducer and activator of transcription 3 (STAT3) is an oncogenic transcription factor that has been implicated in many human cancers and has emerged as an ideal target for cancer therapy. Withaferin A (WFA) is a natural product with promising antiproliferative properties through its association with a number of molecular targets including STAT3. However, the effect of WFA in pediatric neuroblastoma (NB) and its interaction with STAT3 have not been reported. In this study, we found that WFA effectively induces dose-dependent cell death in high-risk and drug-resistant NB as well as multiple myeloma (MM) tumor cells, prevented interleukin-6 (IL-6)–mediated and persistently activated STAT3 phosphorylation at Y705, and blocked the transcriptional activity of STAT3. We further provide computational models that show that WFA binds STAT3 near the Y705 phosphotyrosine residue of the STAT3 Src homology 2 (SH2) domain, suggesting that WFA prevents STAT3 dimer formation similar to BP-1-102, a well-established STAT3 inhibitor. Our findings propose that the antitumor activity of WFA is mediated at least in part through inhibition of STAT3 and provide a rationale for further drug development and clinical use in NB and MM.
Keywords
Introduction
Proteins of the signal transducer and activator of transcription (STAT) family promote tumor proliferation, differentiation, apoptosis, metastasis, invasion, and angiogenesis. In addition, STAT proteins contribute to inflammation and immune response and promote chemo- and radioresistance.1–5 Particularly STAT3 is a key regulator of these processes and is often constitutively activated or overexpressed, thus making this protein a promising target in many solid and hematopoietic human cancers.6,7 STAT3 was initially discovered as a transcription factor induced by interferon-gamma (IFN-γ)8,9 and since it is required for the oncogenic transformation activity of v-src, STAT3 is considered an oncogene. 10 The phosphorylation of STAT3 at residue Y705 triggers STAT3 dimerization, which is required for its translocation to the nucleus where it binds DNA and exhibits its transcriptional activity. A second phosphorylation event at residue S727 is required for optimal transcriptional activity. 11 Aberrant activation of STAT3 can trigger the initiation, development, and progression of human tumors through STAT3-dependent activation of downstream genes that regulate apoptosis, cell cycle, and angiogenesis. 12 Cytokines of the interleukin-6 (IL-6) family, including IL-6, oncostatin M, and leukemia inhibitory factor, activate a number of signaling pathways including STAT3 through JAK1 and JAK2,13,14 which play a major role in the communication between tumor and immune cells. 2
Neuroblastoma (NB) is a pediatric cancer of the sympathetic nervous system and the most common extra-cranial solid malignancy. 15 While significant therapeutic success has been achieved over the past 60 years, the improvement in overall survival rates is mostly limited to low- and intermediate-risk NB patients, while the treatment of aggressive, high-risk NB patients with multiple relapse and refractory disease remains a significant therapeutic challenge. Unlike in most cancers, STAT3 in NB cell lines is typically not constitutively active but can be activated through the influence of the microenvironment, for example IL-6, which is not produced by NB cells but by bone marrow-derived mesenchymal stem cells as well as tumor-associated macrophages. 16 Indeed, elevated levels of IL-6 in the bone marrow and peripheral blood have been linked to poor prognosis in high-risk NB patients and cell culture studies confirmed that IL-6 increased the proliferation and decreased cytotoxic drug-induced apoptosis in response to IL-6-mediated STAT3 activation in NB cells.16,17 Increased and persistent STAT3 activity is commonly detected in hematopoietic malignancies such as multiple myeloma (MM), leukemia, and lymphoma. MM is a cancer of plasma cells that accumulate in the bone marrow and interfere with the production of normal blood cells.
The discovery and isolation of potent anticancer compounds from nature has produced some impressive results in the past, and a respectful number of drugs on the market today are natural products or natural product-inspired derivatives.18,19 Withaferin A (WFA) (Fig. 1) was originally isolated from the plant Withania somnifera and showed antiproliferative properties in several cancer types. A number of potential targets for WFA have been identified (reviewed in) 20 but few have been characterized in more detail and shown to bind directly to WFA. While STAT3 activity inhibition has been investigated in both NB and MM,14,16,21 the antitumor effects of WFA in NB and its impact on STAT3 activity has never been examined. To our knowledge, only one study exists that reported the effect of WFA on nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) in MM. 22
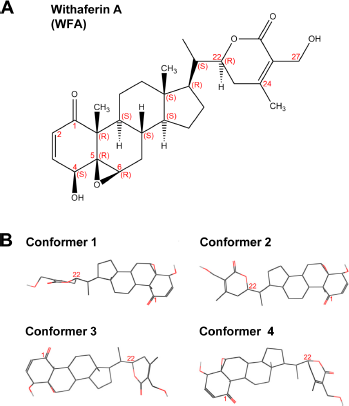
The structure of WFA. (
The present study was designed to test if WFA induces death of NB and MM tumor cells in the presence or absence of IL-6 and to verify if WFA directly binds STAT3. We propose that WFA ablates STAT3 transcriptional activity by preventing dimerization which leads to tumor growth inhibition. This proof-of-concept demonstrates that blockade of STAT3 signaling may be of therapeutic benefit for NB and MM patients.
Experimental Procedures
Mammalian cell cultures and reagents
The human NB cell lines Be(2)-c, SMS-KCNR, and SH-SY5Y were obtained from Dr Giselle Sholler (DeVos Children's Hospital, Grand Rapids, MI). The NB cell line LAN-5 was obtained from Dr Randy Wada (John A. Burns School of Medicine, Honolulu, HI). NB cell line IMR-32 was purchased from American Type Culture (Collection, Manassas, VA). MM cell lines MM1.RL and U266 were obtained from Dr Nancy L. Krett (Northwestern University, Chicago, IL). Cells were maintained in RPMI 1640 (Mediatech Inc, Manassas, VA) containing 10% heat-inactivated fetal bovine serum (FBS) (Atlanta Biologicals, Inc, Lawrenceville, GA), penicillin (100 IU/mL), and streptomycin (100 Ag/mL) (Mediatech). Stock solutions were prepared for WFA at 42.5 mM (Enzo Life Sciences, Inc, Farmingdale, NY) and for S3I-201 at 54.7 mM (Selleck Chemicals, Houston, TX) in dimethyl sulfoxide (DMSO) (Electron Microscopy Sciences, Hatfield, PA). Recombinant human IL-6 (5.0 µg/mL) (PeproTech, Rocky Hill, NJ) was dissolved in 1X phosphate-buffered saline (PBS) and 0.1% bovine serum albumin. WFA, S3I-201, and IL-6 were diluted with culture media before addition to cells. An equal concentration of DMSO was used for control treatments.
Western blot analysis
Lysates from WFA- or S3I-201–treated cells and DMSO-treated (control) cells were prepared using the radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.5], 0.1% [w/v] sodium lauryl sulfate, 0.5% [w/v] sodium deoxycholate, 135 mM NaCl, 1% [v/v] Triton X-100, 10% [v/v] glycerol, 2 mM ethylenediaminetetraacetic acid (EDTA), supplemented with Complete Protease Inhibitor Cocktail, EDTA-free (EMD Millipore, Billerica, MA) and phosphatase inhibitors (20 mM sodium fluoride, and 0.27 mM sodium vanadate). Lysate samples were suspended by rotation at 4°C for a minimum of 30 minutes and clarified by centrifugation for 10 minutes at 15,000 rpm at 4°C. The supernatant was stored at –20°C until further use. Total protein concentration was determined by Bradford dye reagent protein assay (Bio-Rad Laboratories Inc, Richmond, CA). Laemmli buffer (Bio-Rad) containing 10% (v/v) β-mercaptoethanol (EMD Millipore) was added to the lysates and boiled for 5 minutes. Twenty micrograms of total protein was resolved by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene difluoride membranes (Bio-Rad). Membranes were blocked with 5% nonfat dry milk (Santa Cruz Biotechnology, Santa Cruz, CA) in 1X Tris-buffered solution and 0.1% Tween-20 (0.1% T-TBS) for 1 hour at room temperature. The membranes were washed with 0.1% T-TBS and incubated overnight at 4°C with rabbit polyclonal phospho-STAT3 (Y705), rabbit polyclonal phospho-STAT3 (S727), rabbit polyclonal STAT3, or rabbit monoclonal α-tubulin primary antibodies (Cell Signaling Technology, Inc., Danvers, MA). After three 5-minute washes in 0.1% T-TBS, membranes were incubated for 1 hour at room temperature in a fluorescently labeled mixture of secondary goat anti-rabbit antibodies (IRDye 800CW or IRDye 680RD; LI-COR Biosciences, Lincoln, NE). Protein bands were detected using Odyssey Infrared Imaging System (LI-COR).
Cell viability assay
Live/dead cells were determined as follows: cells were cultured in 96-well microtiter plates (Greiner Bio-One Inc, Monroe, NC). All cell lines were suspended in 0.09 mL of medium per well. NB cell lines were seeded at a concentration of 2.0 × 10 4 cells per well, except NB cell line Be(2)-c, which was seeded at 7.0 × 10 3 cells per well. MM cell lines were seeded at 2.5 × 10 4 cells per well. After overnight incubation, cells were treated with increasing concentrations of WFA (0–10 µM) or S3I-201 (0–100 µM) for 48 hours. An equal concentration of DMSO was used as a control. After treatment, 0.5 µg/mL Hoechst 332558 (Biotium, Inc, Hayward, CA) and 0.5 µM TOTO-3 Iodide (Life Technologies, Grand Island, NY) were added in each well and incubated for 10 minutes. Hoechst 332558 is a plasma membrane-per-meant dye that labels all cell nuclei in a population. TOTO-3 Iodide, on the other hand, is a plasma membrane-impermeant dye that specifically labels dying and dead cells. Cell viability was measured with the Operetta High Content Imaging System (PerkinElmer, Waltham, MA) using the image analysis mode “Ready-Made Solution Live/Dead Cell Counting” from the Harmony Database. Briefly, cell count was done for the two subpopulations: live and dead. The threshold was assigned to filter out cells at the dead stain background level. Then, the mode “Select population building block, with the adjusted threshold” was used to identify cells with significant dead cell marker intensity. Finally, the percentage (%) of dead cells in the cell population was calculated [(dead stain positive–-number of objects/nuclei–-number of objects) × 100]. The Microsoft Excel spreadsheet software (Redmund, WA) was used to calculate the mean and the standard error of the mean (SEM) of three independent experiments.
Preparation of nuclear extracts
The preparation of nuclear extracts from cells treated with WFA, S3I-201, or DMSO (control) was carried out with the TransAM STAT3 Transcription Factor Assay kit (Active Motif, Carlsbad, CA), following the manufacturer's protocol. Briefly, MM1.RL and U266 cells (4.0 × 10 6 cells) were seeded in six-well culture plate. Cells were serum-starved (RPMI 1640 media containing 0.1% FBS) overnight and treated with WFA (5 µM) or S3I-201 (100 µM) for 5 hours. After treatment, MM1.RL cells were stimulated with IL-6 (4 ng/mL) for 1 hour. Cells were then washed and collected using ice-cold 1X PBS/PBS supplemented with phosphatase inhibitor buffer. Cells were pelleted by centrifugation for 5 minutes at 300 × g and 4°C. Cells were resuspended in ice-cold hypotonic buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 5 mM sodium fluoride, 10 µM sodium molybdate, and 0.1 mM EDTA) and incubated on ice for 15 minutes. Nonidet P-40 (final concentration of 0.5%) was mixed into the samples and centrifuged for 30 seconds at 4°C. Finally, the nuclear pellet was resuspended in Complete Binding Buffer by rotation at 4°C for 30 minutes and centrifuged for 10 minutes at 14,000 × g at 4°C, saving the supernatant (nuclear cell extract). Protein concentration was determined by Bradford dye reagent protein assay (Bio-Rad).
STAT3 Transcription Factor Assay
STAT3 transcription factor activation was detected and quantified in nuclear cell extracts with the TransAM STAT3 Transcription Factor Assay kit (Active Motif), following the manufacturer's protocol. This kit is based on a DNA-binding enzyme-linked immunosorbent assay (ELISA). In brief, equal amounts of nuclear extracts from cells treated with WFA, S3I-201, or DMSO (control) were added to a 96-stripwell microtiter plate (provided by the kit) to which a STAT3-specific, consensus-binding site oligonucleotide has been immobilized and incubated for 1 hour at room temperature with mild shaking. At the same time a competitive binding control experiment was performed using either wild-type (WT) or mutant (MT) oligonucleotide AM6, provided by the manufacturer. WT oligonucleotide AM6 is a competitor for STAT binding, while the MT oligonucleotide AM6 oligonucleotide has no effect on STAT binding. After incubation, wells were washed three times with wash buffer. A STAT3 antibody which is specific for the epitope on the bound and active form of STAT3 was diluted in Antibody-binding buffer, added to each well, and incubated for 1 hour at room temperature. Afterward, wells were washed and incubated with horseradish peroxidase (HRP)-conjugated antibody for 1 hour at room temperature. Developing solution was then added to each well and incubated for 2–10 minutes at room temperature protected from direct light. The reaction was stopped by adding stop solution. Finally, absorbance was measured at 450 nm using a Synergy Mx Monochromator-Based Multi-Mode Microplate Reader (BioTek Instruments, Inc, Winooski, VT). A nuclear extract from IL-6-stimulated (100 ng/mL) HepG2 cells served as a positive control. Optical density (OD) readings, representing STAT3 transcription factor activation, were calculated and evaluated using the Microsoft Excel spreadsheet software.
Protein-ligand docking
Atomic coordinates from X-ray crystal structures of protein transcription factor STAT3 were retrieved from the Protein Data Bank 23 and used for ligand-docking studies. The nonphosphorylated STAT3 core fragment comprised residues V136-R688 (pdb:3cwgA/3cwgB, dimer) and residues V136-F716 (pdb:4e68A, monomer, in complex with DNA) whereas the Y705 phosphorylated protein consisted of residues V136-F716 (pdb:1bg1A, monomer, in complex with DNA). To expand the coordinates of the monomeric subunits of 1bg1 and 4e68 to their dimer subunits, the coordinates of the dimers were calculated from the BIOMT transformation matrices contained in the pdb files.
WFA (Compound ID: 265237) structure information and descriptive data sets were obtained from the PubChem Substance and Compound Database. 24 Three-dimensional coordinates were available for four diverse conformers, energy minimized by the MMFF94 force field. 25 To explore other conformations in ligand recognition, we also subjected WFA to local energy minimization using the robust Dreiding force field. 26
Docking was carried out to find the possible locations, orientation, and interactions of the WFA-binding sites in STAT3. GRAMM, the original Global Range Molecular Matching method was used on our local host both in high-resolution geometric docking mode27,28 and in low-resolution semi-flexible mode to account for conformational flexibility.29,30 The algorithm predicts the binding pockets by computing the intermo-lecular energy potential in protein–ligand complexes through an exhaustive multidimensional search of translations and rotations of the components and using correlation techniques with Fast Fourier transform.
The docking simulations were run with every possible permutation of the phosphorylated and nonphosphorylated monomers and dimers in complexes with the four canonical and the local energy-minimized WFA conformers, and in the presence and absence of bound DNA as the available three-dimensional data permitted. Altogether 100 sets (50 geometric and 50 semi-flexible) were screened for the binding sites and the first 20 binding locations of each set were scored by the number of occurrences of residues in the contact areas among the various protein–ligand pairs. The spatially most invariant complexes were selected as the most probable models. The predicted binding sites were visualized and analyzed with the ICM-Browser (Molsoft LLC, San Diego, CA, USA). Chemical structure drawing was performed employing the ICM Molecular Editor.
Results
WFA induces dose-dependent cell death in NB and MM
To investigate if WFA induces cell death in cell cultures of NB and MM, we tested increasing drug concentrations against a panel of tumor cell lines. To our knowledge, WFA has not been studied in NB, and only one report was found evaluating WFA in MM cells. 22 As shown in Figure 2, WFA induced cell death in seven tumor cell lines in a dose-dependent manner between 0 and 10 µM, with inhibitory activity detected as low as 0.625 µM, in IMR-32 NB cells. At 10 µM, cell death was significant in all tested tumor cell lines and ranged from ~40% to 90%, depending on individual lines (Fig. 2A). Importantly, WFA also impeded the growth of chemotherapy treatment-resistant Be(2)-c, SMS-KCNR, and MM1.RL tumor cells as well as of U266 cells harboring persistently activated STAT3. For direct comparison, the STAT3 inhibitor S3I-201 (see Supplementary Fig. 1) was used and revealed that about 10-fold higher drug concentrations were required to achieve comparable results, under identical experimental conditions (Fig. 2B). Furthermore, S3I-201 did not exert any antiproliferative effects on chemotherapy-resistant MM1.RL cells, even at the highest drug concentrations (100 µM) while WFA induced about 50% cell death in this cell line at 10 µM.
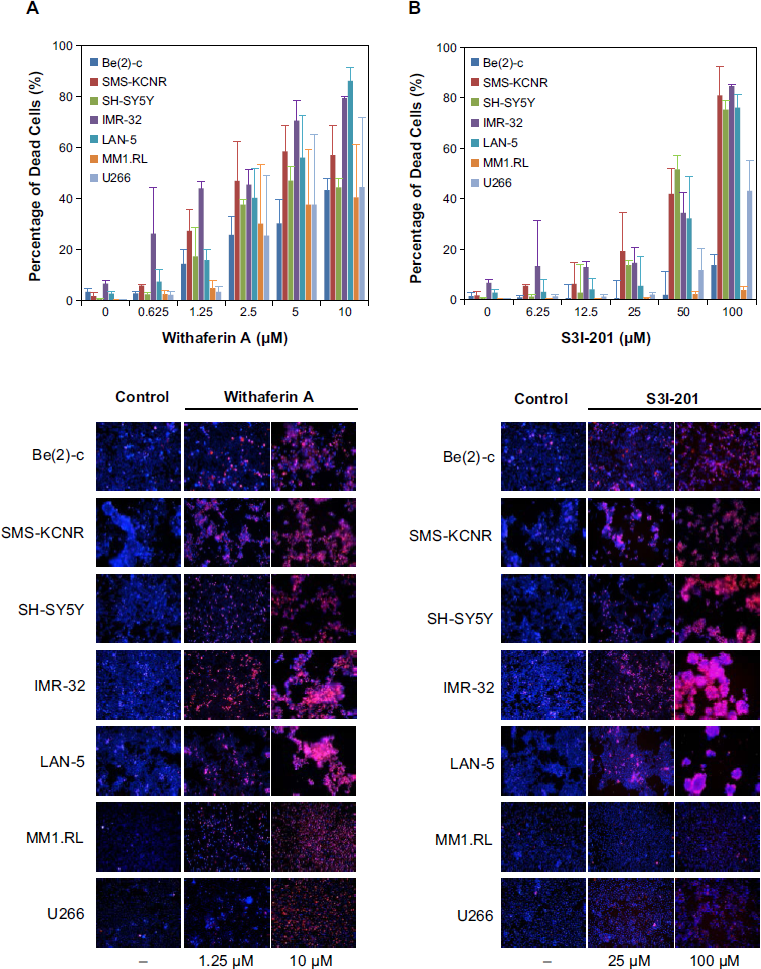
WFA induces cell death in NB and MM. Cell viability analysis of NB cell lines (Be(2)-c, SMS-KCNR, SH-SY5Y, IMR-32, and LAN-5) and MM cell lines (MM1.RL and U266) treated with increasing concentrations of (
Additional studies were performed at the National Cancer Institute (NCI) with the standard NCI 60 cell line panel and confirmed that WFA exhibits broad-range, dose-dependent potency in many tumor types including leukemia, non-small cell lung cancer, colon cancer, central nervous system (CNS) cancer, melanoma, ovarian cancer, renal cancer, prostate cancer, and breast cancer (see Supplementary Fig. 2), thus rendering WFA an interesting anticancer agent with broad application potential.
WFA suppresses STAT3 phosphorylation in NB and MM
Because WFA binds to the protein near the Y705 phos-pho-tyrosine residue of the STAT3 Src homology 2 (SH2) domain and prevents STAT3 dimer formation in silico (Fig. 3), we examined whether WFA interferes with STAT3 phosphorylation at Y705 in vivo, thus providing a possible mechanism for the observed induction of cell death (Fig. 2). Since phosphorylation at S727 enhances STAT3 transcriptional regulatory activities, 31 we tested both phosphorylation sites. STAT3 inhibitor S3I-201 was included as control for direct comparison. In NB cells, the phosphorylation of STAT3 at Y705 was detected in IMR-32 and LAN-5 control cells and was reduced in response to treatment with WFA and S3I-201 (Fig. 3A). Phosphorylation of STAT3 at S727 was only observed in LAN-5 cells. In MM cells, phosphorylation of STAT3 at Y705 and S727 was significantly reduced compared to U266 control cells, which expresses persistently activated STAT3 (Fig. 3B). In some cell lines, total STAT3 levels also decreased in the presence of S3I-201 and/or WFA, suggesting that both inhibitors exert STAT3 phosphorylation-indepen-dent effects that induce cell death. In cell lines where tubulin was reduced, a more general cell death mechanism might be triggered in response to an overall protein shutdown.
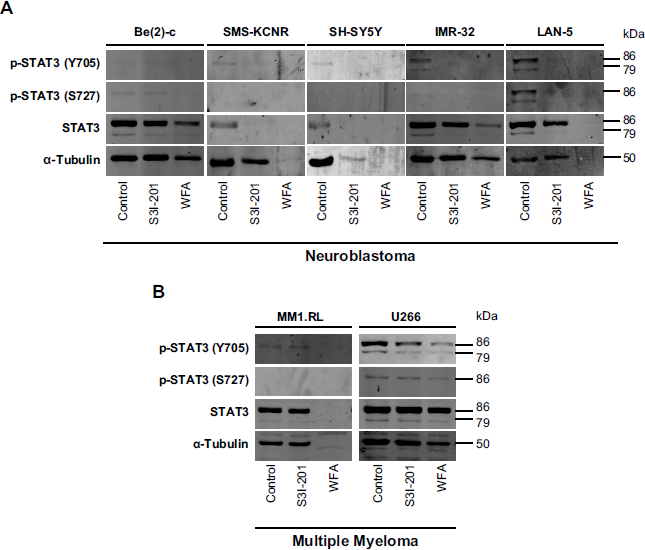
Effect of WFA on STAT3 phosphorylation in NB and MM. Western blot analysis of p-STAT3 (Y705 and S727) and total STAT3 from whole-cell lysates of (
WFA inhibits IL-6–mediated and persistent STAT3 activation in MM cells
Since IL-6 induction is known to trigger STAT3 activation in MM cells, we treated MM1.RL cells with WFA at various time points, followed by IL-6 treatment for 1 hour. IL-6 strongly induced STAT3 phosphorylation at Y705 but not S727 and was inhibited by WFA after only 2 hours of incubation (Fig. 4A); however, total STAT3 levels were reduced after exposure of cells to WFA for more than 4 hours, suggesting that WFA also downregulates STAT3 protein if cells are exposed to the drug for longer time periods. In persistently activated U266 cells, WFA reduced STAT3 phosphorylation in a time-dependent manner without significantly altering total STAT3 levels up to 6 hours (Fig. 4B). Total STAT3 levels declined at drug exposure times beyond 10 hours. WFA efficiently blocked STAT3 phosphorylation at Y705 in both nuclear and cytosolic extracts of IL-6–induced MM1.RL and persistently activated U266 cells. Total STAT3 protein levels declined in the cytosolic extracts but stayed the same or slightly increased in the nuclear extracts, after WFA treatment (Fig. 4C).
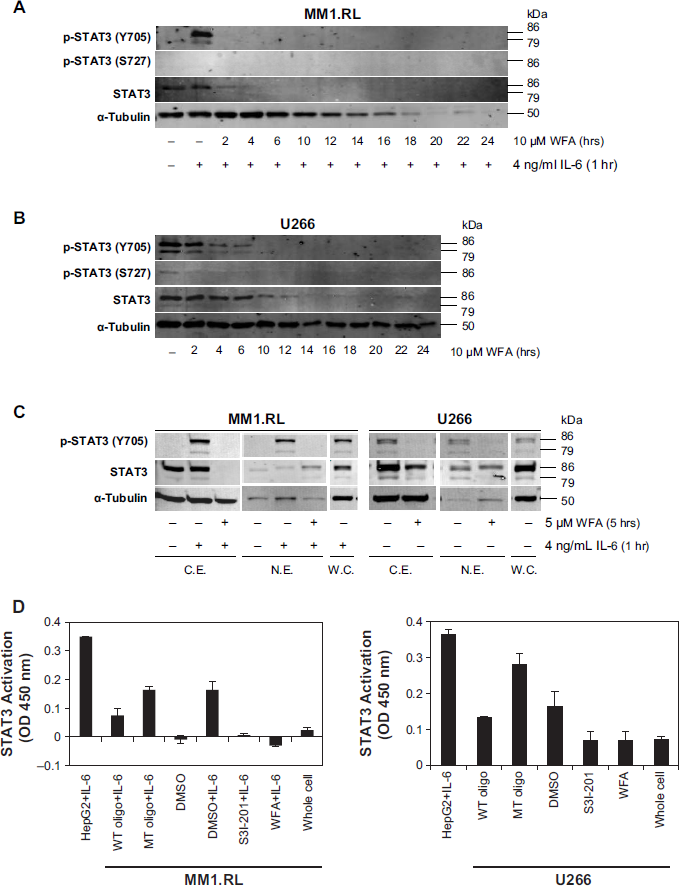
Effect of WFA on constitutive and IL-6–induced STAT3 transcriptional activation and translocation in MM. Western blot analysis of p-STAT3 (Y705 and S727) and total STAT3 from whole-cell lysates of (
Summary of docking results with WFA.
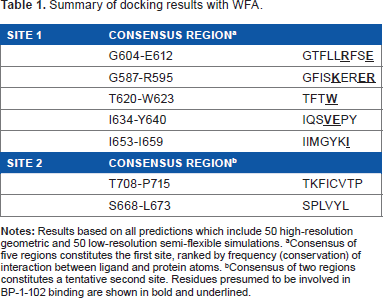
Consensus of five regions constitutes the first site, ranked by frequency (conservation) of interaction between ligand and protein atoms.
Consensus of two regions constitutes a tentative second site. Residues presumed to be involved in BP-1-102 binding are shown in bold and underlined.
Next we examined the effect of WFA on the transcriptional activity of STAT3 using a highly sensitive DNA-binding ELISA assay that measures STAT3 transcription factor activation. The STAT3 transcriptional activity in nuclear extracts of IL-6-induced MM1.RL cells and persistently activated U266 cells was strongly inhibited by WFA at 5 µM which was comparable to effects achieved by STAT3 inhibitor S3I-201 at 100 µM (Fig. 4D).
Computational modeling and docking of WFA into STAT3
To examine if WFA binds STAT3, we performed extensive computational modeling and docking simulations. WFA, a potential anticancer withanolide, is a cell-permeable steroidal lactone with a modified ergostane framework whose molecular structure is shown in Figure 1A. It has four distinct canonical conformers separated by energy barriers higher than 2 kcal/mol (Fig. 1B). Each conformer was individually used in the docking simulations. We also tested a fifth conformer (generated by local energy minimization), which represented the approximate average geometry of the canonical conformers.
Three STAT3 crystal structures are available, one phosphorylated (1BG1) and one nonphosphorylated (4E68) structure, each in complex with DNA, and one nonphosphorylated structure for the unbound protein (3CWG). The STAT3 core protein encompasses an N-terminal 4-helix coiled-coil structure (residues V136-A320), a DNA-binding domain (A406-S514), a structurally conserved SH2 domain also frequently found in oncoproteins and intracellular signal-transducing proteins (T600-R688), and a C-terminal acidic region containing phosphorylation sites Y705 and S727 (the latter site is not present in the X-ray structure). STAT3 forms a biologically active homodimer and we explored possible binding modes both for the monomers and the dimers. A total of 100 different docking scenarios were screened using all possible combinations of the five ligand conformers and the various protein receptors (monomer/dimer, phosphorylated/nonphosphorylated protein, with/without DNA). The docking calculations were performed on each scenario by geometric complementarity and semi-flexible docking to take into account inherent receptor flexibility. From each run, the 20 lowest energy-docking positions were retained which yielded a total of 2000 poses. The presumed WFA-binding sites were ranked by conservation score, ie, the frequency of occurrence of a residue in a contact area. The contact area was defined as that consisting of the residues within a radius of 5 Å centered at the ligand.
Based on the conservation scores of all the residues, we identified two main binding locations situated at adjacent areas of the SH2 domain. A consensus of five regions constituted the first site comprising residues G604-E612 (Rank 1), G587-R595 (Rank 2), T620-W623 (Rank 3), I634-Y640 (Rank 4), and I653-I659 (Rank 5), which formed the sub-pockets of the WFA contact areas (Table 1). The absence and presence of DNA does not affect the location of the binding sites despite the fact that the dimer adopts a distinctly different, locally more constrained conformation upon complex formation with DNA. Figures 5 and 6 show the binding of WFA to phosphorylated and nonphosphorylated STAT3 dimer, in the presence and absence of complexed DNA, respectively. Site 1 is located in a water-accessible surface depression of the SH2 domain close to the dimerization interface of STAT3 and is essentially the same in all structures tested. Both chains were found to simultaneously bind ligands in the homodimer. Y705 was reported earlier to interact with dimerization-disrupting small molecules such as S3I-201.1066 32 (also referred to as S3I-201), and WFA bound to Site 1 appears to sit within 5 Åx from Y705 of the neighboring chain of the dimer near to the dimerization interface. Site 1 overlaps but is not necessarily entirely identical to the site for interaction with BP-1-102 (see Supplementary Fig. 1), an SH2 domain ligand that inhibits STAT3 activation and functions in vitro and in vivo. 33 Residues K591, E594, R595, R609, E612, W623, I634, V637, E638, and I659 were implicated in computational modeling of BP-1-102 binding, and all these residues are also present in the ranked sub-pockets of Site 1 (Fig. 7).
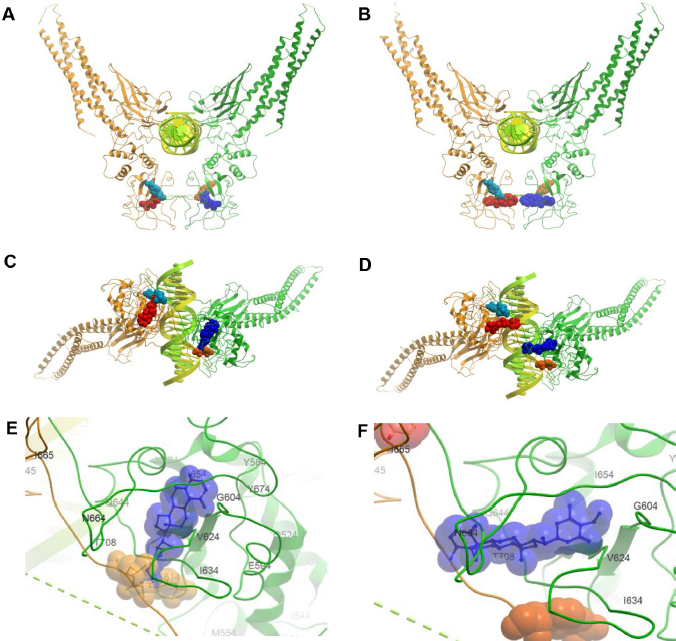
Binding of WFA (Conformer 1) to Y705-phosphorylated STAT3 dimer in complex with DNA (1BG1:V136-F716). Both chains bind ligands independently. (
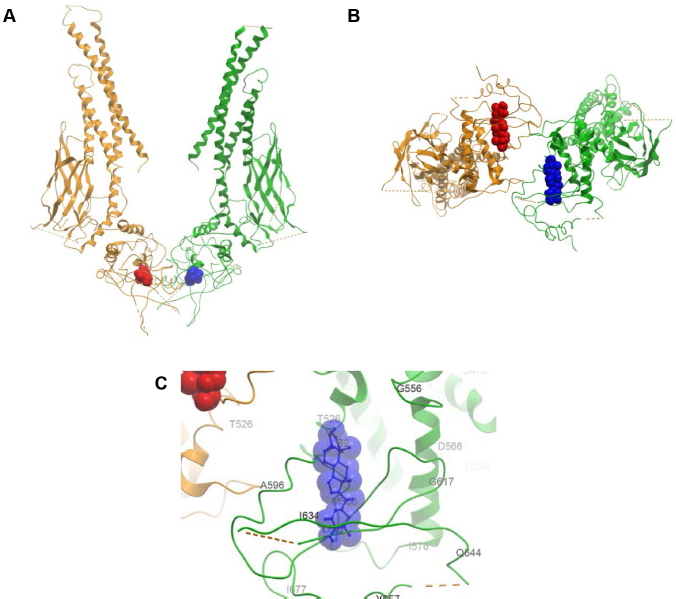
Binding of WFA (Conformer 2) to nonphosphorylated STAT3 dimer (3CWG:V136-R688). (
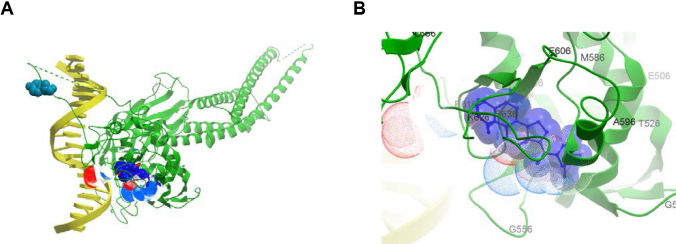
Binding of WFA (Conformer 3) to nonphosphorylated STAT3 monomer in complex with DNA (4E68:V136-F716). (
The second site covers a broader, more open binding cleft and involves residues T708-P715 (Rank 6) and S668-L673 (Rank 7) (Table 1). It is located near the key site of phosphorylation, Y705 from both chains of the dimer (Fig. 6). While Site 1 was present in all combinations of the phosphorylated receptor-ligand pairs, it was largely absent or its location altered in the nonphosphorylated protein. Furthermore, we found that Conformer 1 could dock in head and tail orientation at Site 2. The O1 orientation toward the dimerization interface appeared more favorable as it occurred with two-times higher frequency than the reversed position. Sub-pocket 6 is situated in the immediate vicinity of Y705 of the same chain and the proximity of Y705 to WFA was also observed in a variety of slightly different binding poses. At any rate, WFA binding results in a thermodynamically stable structure that leads the dimer to undergo a transition.
Discussion
In this study, we performed a series of cell culture-based studies with five NB and two MM tumor cell lines and measured cell proliferation as well as STAT3 phosphorylation and STAT3 transcriptional activation patterns, in the presence or absence of IL-6 or in cells with constitutively active STAT3. Our data showed that WFA induced cell death in a dose-dependent manner at significantly lower drug concentrations when compared with the STAT3 inhibitor S3I-201. At 5.0 µM, WFA induced ~50% cell death in most cell lines while S3I-201 at similar concentration had almost no inhibitory potential. Strikingly, cell lines IMR-32 and Be(2)-c, which are MYCN-amplified and more aggressive NB subtypes, were killed most effectively with WFA. While the inhibitory effects on MM cell lines were less pronounced, WFA at higher concentrations (10 µM) inhibited both MM1. RL and U266 lines equally at about 50%. In contrast, S3I-201 only inhibited U266, but not MM1.RL cells, thus suggesting that WFA is also effective in a drug-resistant cell type that did not respond to S3I-201.
STAT3 phosphorylation at Y705 of untreated and nonin-duced (control) NB cells was observed in IMR-32 and LAN-5 but not in Be(2)-c cells and only weakly in SMS-KCNR and SH-SY5Y cells. It is possible that of the five NB cell lines tested, IMR-32 and LAN-5 express persistently activated STAT3, similar to U266 cells. Of note, IMR-32 and LAN-5 were the two cell lines most potently inhibited by WFA and S3I-201, suggesting that STAT3 activity is important in those cell lines and contributes to cell proliferation, and cell death is effectively induced by both STAT3 inhibitors (Fig. 2). WFA successfully inhibited STAT3 phosphorylation in both IL-6-induced and persistently activated MM cell lines in a time-dependent manner. However, WFA also impacted STAT3 and tubulin total protein levels as well as GAPDH protein levels (not shown) to various degrees, especially after longer (>10 hours) exposures, suggesting additional inhibitory mechanisms at play that kill cells and are triggered in response to WFA and S3I-201 treatments. Interestingly, subcellular separation experiments with cytosolic and nuclear extracts of either IL-6–activated MM1.RL or persistently activated U266 cells revealed that WFA inhibited the phosphorylation of STAT3 at Y705 in both fractions but total STAT3 levels declined in the cytosolic fraction and remained consistent or slightly increased in the nuclear fraction after WFA treatment. This was unexpected since it is known that the inhibition of STAT3 phosphorylation at Y705 prevents STAT3 dimerization which blocks the subsequent transport of STAT3 to the nucleus. Our result suggests that despite the expected absence of phosphorylated STAT3 in the nucleus, a fraction of nonphosphorylated STAT3 is present in the nucleus after WFA treatment. It is possible that either nonphosphorylated STAT3 is transported to the nucleus and/or WFA is transported to the nucleus and prevents the phosphorylation of STAT3 in the nucleus. Regardless, our experiments with nuclear cell extracts showed that the transcriptional activity of STAT3 is clearly inhibited by WFA using a highly sensitive, quantitative STAT3 transcription factor ELISA (Fig. 4D). Of note, WFA inhibited STAT3 transcriptional activity to the same degree at a 20-fold lower concentration compared to STAT3 inhibitor S3I-201, thus proposing at least equal target specificity and higher drug potency.
While STAT3 inhibition by WFA has been reported in other cancer cell systems,34–36 those findings did not reveal whether the inhibition of STAT3 activity was due to a direct binding of WFA with STAT3. To investigate this, we performed several WFA-STAT3-docking simulations in silico and report structure-based evidence in support of a direct molecular interaction between WFA and STAT3. Furthermore, we revealed several key reaction sites of WFA that largely overlap with the well-characterized STAT3 inhibitor BP-1-102. 33
We first performed computational docking simulations to predict the binding site(s) of WFA on STAT3. Our results showed that two binding interfaces may exist: one primary binding site (Site 1) in the SH2 protein domain with high probability and a tentative secondary site (Site 2) in its neighborhood with low probability. SH2 domains typically function as phosphorylated tyrosine recognition domains on other proteins thus playing a fundamental role in signal transduction. They function as regulatory modules of phosphorylation-dependent protein-protein interactions inducing changes in gene expression and other cellular processes. The contact areas of Site 1 include Y705, the site of tyrosine phosphorylation from the adjoining chain of the protein dimer. Site 2 is located in close proximity to Y705 of the same chain and may come proximate to Y705 from the other chain if binding is accompanied by a conformational change. Thus, binding of WFA can directly affect phosphorylation and coupled signal transduction at any of these two sites via changes in physical-chemical factors such as charge, hydrogen bonding, solvent accessibility, and side-chain conformations. Ligand reorganization may also play an important role in interacting with Y705 and in determining the stability of the STAT3 complex with the ligand as all conformers of WFA appeared to bind to the protein with decreasing order of occurrence from Conformer 1 to Conformer 4. It is plausible that the distinctly different conformers undergo a structural transition upon binding, resulting in a single active conformer state that provides the highest stability for the complex.
A further and more likely mechanism is dimer disruption. Both sites lie on the same interface of the STAT3 dimer where they can interrupt or alter dimer formation and thereby inhibit normal STAT3 functions similarly to other small-molecule inhibitors such as BP-1-102 and S3I-201 (see Supplementary Fig. 1). One may speculate that WFA prevents the two protein chains from moving into closer proximity upon ligand binding and that it would dissociate preexisting dimers, thereby arresting the activity of STAT3, as observed in experiments depicted in Figure 4D.
Molecular docking further revealed that both chains of the dimer can bind ligands simultaneously. However, it is not known if sites on both chains must be occupied at the same time or binding to only one chain is enough for inhibition of protein function. Since the sites are relatively close to each other and are close to the dimer interface, there is a possibility that binding at only one site is sufficient. Binding may result in some conformational change rendering the site on the other chain inaccessible, or the two chains may actually form one unified binding pocket upon a conformational change unless the dimer dissociates.
Site 2 displayed a level of heterogeneity in binding. The most probable low-energy binding mode could be determined only by scoring and was not obvious from visual inspection. This site is not as tight as the primary site as it allows Conformer 1 to dock in two opposing orientations. This is not entirely surprising given that Conformer 1 is a relatively “flat” molecule and both its head (O1) and tail (O6) contain a conjugated O=C–C=C element and the head/tail moiety can rotate relative to each other.
It is important to point out that there are small structural variations and missing residues in the vicinity of Site 2 in the X-ray structures, which may cause docking simulations to give unreliable or spurious results. Therefore, while the location of Site 2 near Y705 is an attractive hypothesis, further investigation is needed and at this point one may consider it as a tentative possibility.
WFA is one of the most bioactive compounds known to exert antiproliferative, proapoptotic, and also anti-invasive and antiangiogenic effects. It has been associated with a number of molecular targets, for example, the proteasome,37–40 Notch-1, 41 NF-κB,41,42 cell division cycle 37/ heat shock protein 90 (Cdc37/HSP90), 43 reactive oxygen species (ROS),44–46 cyclooxygenase-2 (COX-2),35,46,47 p38 mitogen-activated protein kinase (p38 MAP kinase),48,49 AKT/protein kinase B (PKB),41,50 estrogen receptor alpha (ER-alpha), 51 vimentin, 52 vascular endothelial growth factor (VEGF), 53 and acetyl and butyryl cholinesterase. 54 Molecular insights into many of these multifunctional activities of WFA and its potential therapeutic effects were recently reviewed. 20 A wide range of targets and the resulting pleiotropic effects are not uncommon in natural product research, as exemplified by resveratrol.55,56 Furthermore, a number of dietary compounds from nature including capsa-icin and curcumin have been proposed to interfere with the STAT regulatory system. 21
However, whether all those reported targets interact directly with WFA or are secondary outcomes remains largely unknown. For example, it was suggested that the proteasome is a primary target for WFA;38–40 however, attempts to determine the 20S proteasome:WFA complex structure did not result in any defined electron density for the natural product (data not shown, Michael Groll, Technical University of Munich, Germany; personal communication). In contrast, we provide in this study first evidence that STAT3 is a direct target for WFA, which binds in a manner similar to BP-1-102, a potent STAT3 inhibitor (Fig. 7), thus strongly supporting that WFA is a STAT3 inhibitor that may be further developed into a useful anticancer therapeutic.
Author Contributions
Carried out cell viability, immunofluorescence, and Western blot studies, assembled corresponding figures, and helped draft the manuscript: LPY. Carried out the computational model analyses, assembled corresponding figures, and wrote the computational sections of the manuscript: GM. Carried out the nuclear fractionation and STAT3 transcription factor assays and assembled corresponding figures: JOA. Conceived and directed the study, received funds for study, participated in the design of experiments, and wrote the manuscript: ASB. All authors read and approved of the final version of the manuscript.
Supplementary Materials
Supplementary figure 1
Small molecule inhibitors of STAT3. (
Supplementary figure 2
Antiproliferative effect of Withaferin A (WFA) in the NCI 60 cell line screen. (
Footnotes
Acknowledgments
We thank Dr Giselle Sholler (Helen DeVos Children's Hospital, Grand Rapids, MI) for providing NB cell lines Be(2)-c, SMS-KCNR, and SH-SY5Y, Dr Randal Wada (University of Hawaii at Manoa, Honolulu, HI) for NB cell line LAN-5, and Dr Nancy L. Krett (Northwestern University, Chicago, IL) for MM cell lines MM1.RL and U266. The NCI is thanked for providing the NCI-60 cell line panel data for WFA.