
Abstract
Rheumatoid arthritis (RA) is a progressive autoimmune disease characterized by chronic synovitis and articular destruction. Pain is the earliest and most important symptom. The Janus kinase/stat signaling pathway not only participates in the physiological processes such as the growth and differentiation of normal cells, but also plays a significant role in the pathological mechanisms such as pain in RA patients. Pain in RA patients is mediated by both inflammatory and non-inflammatory factors, including central sensitization and peripheral sensitization. Cytokines can regulate the nociceptor threshold through the JAK pathway, which leads to sensitization. In this review, we provide an overview of the physiological basis of pain modulation, the underlying importance of cytokines and JAK/STAT pathway in pain modulation, and finally introduce the performance of JAK inhibitors in clinical research. Having a better understanding of the mechanism of pain in RA may provide new therapeutic ideas and directions for the clinical improvement of pain in RA patients.
Keywords
Introduction
Pain represents the predominant and pervasive hallmark of rheumatic diseases, serving as the primary impetus for patients seeking healthcare professionals’ intervention. 1 A survey encompassing 1204 rheumatoid arthritis (RA) patients unveiled that 68.6% identified pain as the utmost symptom necessitating amelioration. 2 In recent years, pain has transcended its isolated symptom status and emerged as a multidimensional entity, often accompanied by psychological distress, sleep disturbances, and compromised social function. 3 While joint pain has conventionally been attributed solely to inflammation, a plethora of studies have demonstrated that RA patients may persistently experience pain even when inflammation is effectively controlled. 4 Importantly, pain occurrence is intricately linked not only to inflammatory factors but also to a range of non-inflammatory influences. 5 Pain significantly impacts patients’ quality of life and serves as a crucial determinant in assessing disease activity. Clinical evaluations of disease activity consistently incorporate at least one pain assessment measure. Moreover, improvement in pain represents a decisive criterion for evaluating the efficacy of clinical interventions and guiding medication plan adjustments. Consequently, gaining a comprehensive understanding of pain-related mechanisms not only enhances patients’ quality of life but also has a positive impact on the development of novel analgesic targets. In this review, we focus on investigating the inflammatory and non-inflammatory mechanisms underlying pain occurrence in RA patients, with a specific emphasis on elucidating the role of the JAK pathway in pain modulation. Additionally, we perform a rigorous analysis of clinical studies to elucidate the effects of JAK inhibitors on pain regulation in RA patients.
Pain and rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic multisystem autoimmune disease accompanied by synovitis, which is mainly characterized by erosive and symmetric polyarthritis. 6 The basic pathological changes are chronic inflammation of joint synovium and pannus formation, which may develop into joint deformity and loss of function in the later stages. Arthralgia is often the first symptom and the main reason for patients to seek medical treatment. Most of the pain is symmetrical and persistent with tenderness in RA. Joint swelling, tenderness, and structural destruction can lead to joint movement disorders. 7 In the early stage of disease activity, the pathological mechanism of RA pain can be attributed to the increase of inflammatory effectors. However, as the disease progresses, the pain of RA can also be caused by non-inflammatory factors, including bone damage and nociceptive sensitization. 8 In addition, mood is also one of the important factors affecting RA pain. 9 A good mood is beneficial to the improvement of joint pain, while anxiety and tension often aggravate the disease. Negative emotions, especially anxiety, and depression, can affect the subjective perception of pain in RA patients, enhance the processing of pain in the nervous system, and then aggravate the sensation of pain. 10 In addition, sleep disorders can also affect the pain perception of patients, and even create a vicious cycle that affects the normal life of patients. Pain is not only a burden in the work and life of patients, but also one of the social and economic burdens. Therefore, there is an urgent need to improve our understanding of the physiological mechanism of RA pain in order to find a more effective clinical treatment that can relieve pain.
The physiological basis of pain modulation
The burden of pain on patients and society is immeasurable. Chronic pain, as defined by the International Association for the Study of Pain, refers to unpleasant sensory and emotional experiences associated with actual or potential tissue damage. 11 The perception of pain relies on nociceptive receptors, which are specialized sensory fibers that respond to high-intensity mechanical, chemical, or thermal stimuli that may cause tissue damage. Nociceptors encompass myelinated nerve fibers A and unmyelinated nerve fibers C, terminating in the skin, muscles, joints, and visceral organs, with their cell bodies located in the dorsal root ganglion (DRG) and the trigeminal nerve. 12 These conductive nerve fibers typically have high thresholds and only generate excitation in response to noxious stimuli, thus earning the term “nociceptors”. In the case of joints, most structures, such as the synovium and joint capsule, are primarily innervated by peripheral afferent fibers of the dorsal root ganglion. Nociceptor activation occurs when the stimulus reaches a range that poses a danger of injury. Studies have revealed a significant increase in nerve fibers in the synovial membrane of joints in mouse arthritis models induced by complete Freund’s adjuvant, suggesting that these nerve fibers may be the primary cause of joint pain. 13 During joint inflammation, resident cells and immune cells within the joint can release proinflammatory cytokines. The synovial inflammatory response contributes to the activation or sensitization of pain-afferent fibers. Subsequently, pain signals are transmitted to the spinal dorsal horn and further to the thalamus and higher processing centers, including the somatosensory, insular, and cingulate cortices, as well as the reticular and limbic systems. 14
The generation of chronic pain is associated with inflammatory factors as well as non-inflammatory factors. Although the patient’s pain correlates with the level of inflammation, the level of inflammation does not show a direct relationship with the level of joint pain. In a large proportion of patients with rheumatoid arthritis, pain persists even when the level of disease activity continues to be relieved or at a minimum. 15 A study by William et al. found that although patients were treated with DMARDs in the early stage, the pain of some patients was not completely improved, and even 27% of patients had an increase in pain compared with baseline. 16 It is reasonable to assume that the occurrence of pain is also related to non-inflammatory factors, including damage to joint structures and abnormal neuronal activity. In other words, there are patients for whom the pain is primarily caused by factors other than inflammation and for whom intensive use of DMARDs may not be effective. 8 Leffler has provided evidence for the involvement of the central nervous system(CNS) in pain regulation through the mechanism of Diffuse Noxious Inhibitory Controls (DNIC). 17 DNIC functions as an autonomic regulatory system that attenuates the transmission of pain signals by concurrently or sequentially administering noxious stimuli to different anatomical regions. In the context of rheumatoid arthritis, this study identified impaired DNIC functionality in affected patients. Typically, the receipt of diffuse noxious stimuli triggers an inhibitory response within the central nervous system, thereby leading to a diminished perception of pain. Conversely, individuals with rheumatoid arthritis exhibited diminished or absent pain inhibitory responses, indicating an inability of the central nervous system to adequately suppress pain signals. Consequently, these patients experience heightened sensitivity towards painful stimuli and a reduced pain threshold. These findings substantiate the pivotal role of the central nervous system in modulating pain perception and offer valuable insights into potential mechanisms underlying the heightened pain experienced by individuals with rheumatoid arthritis. 17 Regulation by the CNS through the central nervous system can either promote pain or suppress it. There are three main mechanisms by which the central nervous system regulates pain: First, the descending facilitatory pathways; Second, the descending inhibitory pain pathways; Third, central sensitization. 5 The descending inhibitory pathway and descending facilitation pathway have similar mechanisms of action, mainly relying on such neurotransmitters as serotonin and norepinephrine. These transmitters can act on the periaqueductal gray matter, nucleus raphe magnus, and the rostral ventromedial medulla, sending signals down from the brain to the spinal cord. 18
The third mechanism of pain regulation is central sensitization. Central sensitization refers to abnormalities in the pain processing within the spinal cord and brain, leading to increased pain sensitivity in widespread areas and an enhanced overall response state of the central nervous system to sensory impulses. This phenomenon manifests as diffuse hyperalgesia and allodynia. Diffuse hyperalgesia refers to an excessively strong sensation of normally painful stimuli, while allodynia is the perception of pain in response to non-painful stimuli. 8 The development of neuronal sensitization involves the active participation of inflammatory mediators. During acute inflammation, infiltrating inflammatory cells and resident cells in the joint sensitize neurons by releasing inflammatory mediators, such as bradykinin, prostaglandins, and proinflammatory cytokines like tumor necrosis factor. These substances further enhance sensory conduction and neuronal excitability. Mechanistically, hyperalgesia is caused by increased sensitivity and/or expression of channels involved in mechanical or thermal stimuli, including ion channels and voltage-gated ion channels like transient receptor potential vanilloid 1 (TRPV1), which affect the activity of sensory neurons. TRPV1 is a non-selective cation channel, 19 and its activation by thermal stimulation results in ion influx, converting the thermal stimulus into potential conduction. Sensitization of TRPV1 leads to its opening at lower temperatures than normal, thus reducing the threshold for hyperalgesia. 20
Roles of cytokines and JAK pathways in pain modulation
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway has emerged as a pivotal intracellular signaling pathway involved in cytokine receptor signaling, garnering significant attention in research. It plays a crucial role in cell survival, growth, development, and differentiation, particularly in the realms of immunity and hematopoiesis. 6 The JAK family comprises four members: JAK1, JAK2, JAK3, and TYK2, each selectively binding to different ligands. 21 When cytokines bind to their receptors, JAK tyrosine kinases are activated and transmit regulatory signals.
These cell ligands range widely, including some cytokines, hormones, colony-stimulating factors. Upon binding to specific cell surface receptors, cytokines induce conformational changes that trigger the activation of JAK kinases. These activated JAK kinases, in turn, phosphorylate downstream signaling molecules, orchestrating cellular responses. Another group of JAK pathway modulators encompasses growth factors, including erythropoietin and granulocyte colony-stimulating factor (G-CSF). Binding to their respective receptors, these growth factors initiate JAK activation, culminating in a cascade of phosphorylation events that mediate cellular signaling. In addition, there are also some hormones that can exert their influence on the JAK pathway as ligands. For instance, growth hormone (GH) binds to its receptor, initiating JAK activation and subsequent phosphorylation of transcription factors. This phosphorylation cascade regulates gene expression and contributes to GH-induced growth effects.
In addition, the JAK pathway can be pharmacologically targeted by specific drugs. Janus kinase inhibitors (JAK inhibitors) represent a class of therapeutics that directly bind to the active site of JAK kinases, effectively blocking their activity. By inhibiting downstream phosphorylation events, JAK inhibitors modulate the JAK pathway and hold promise for therapeutic intervention.
Upon JAK-mediated phosphorylation, STATs translocate to the nucleus as homodimers or heterodimers, binding to DNA and regulating gene expression (Figure 1). Currently, seven mammalian STAT family members have been identified, namely STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. Specific STATs are activated by different cytokines, initiating transcription and upregulating the expression of cytokines and other immune components. Positive feedback loops can also form within this network. Numerous animal studies
22
investigating pain have directly associated alterations in the JAK/STAT pathway with pain regulation. For instance, spinal cord injury rapidly activates the JAK/STAT3 pathway in spinal dorsal horn microglia, concurrent with increased IL-6 levels in the spinal cord. Therefore, the study of the analgesic effect of the JAK/STAT3 pathway is intricately intertwined with cytokine investigations. The activation of the JAK/STAT signaling pathway involves the following steps: (1) Binding of cytokines or growth factors to their corresponding receptors, leading to receptor dimerization and recruitment of associated JAK kinases. (2) Activation of JAK kinases, resulting in receptor tyrosine phosphorylation and formation of STAT docking sites. (3) Phosphorylation of STAT proteins on tyrosine residues. (4) Dissociation of STATs from the receptor, forming either homodimers or heterodimers. (5) Translocation of STAT dimers into the nucleus, where they bind to DNA and regulate transcription.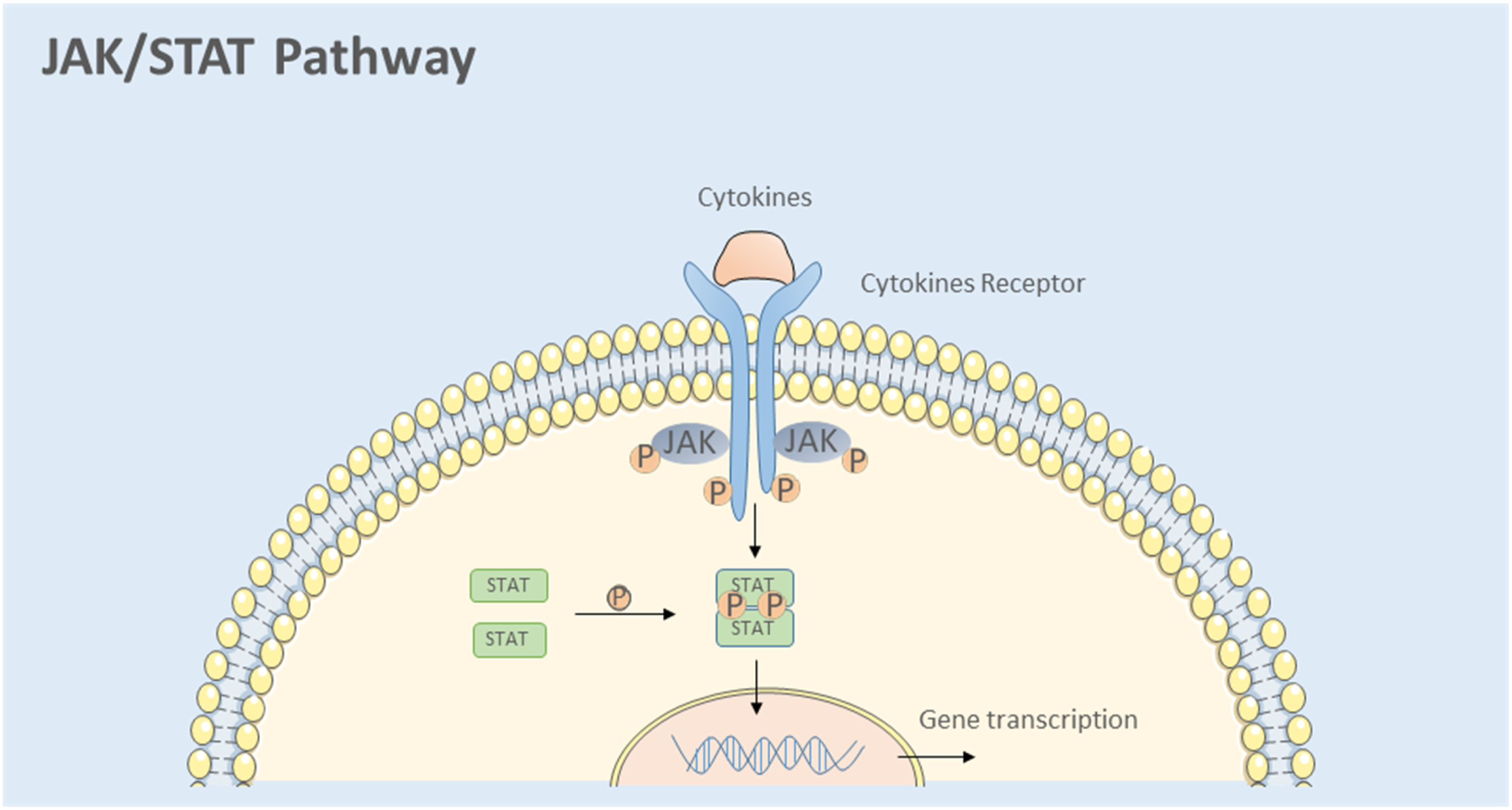
Cytokines involved in the pathogenesis of RA.

The Janus Kinase (JAK)/Signal Transducer and Activator of Transcription (STAT) pathway regulates cytokine-mediated pain in rheumatoid arthritis (RA). Certain cytokines associated with RA pain, such as Interleukin (IL)-1β, IL-6, IL-10, and Tumor Necrosis Factor (TNF)-α, can activate different JAK proteins, leading to the phosphorylation of respective STATs and modulation of gene transcription. For instance, IL-17 activates JAK2 pathway, phosphorylates STAT1 and STAT3, thereby inducing the production of effector proteins. ↑: The cytokines that enhance responsiveness to mechanical pain and increase nociception. ↓: The cytokines that reduce responsiveness to mechanical pain and alleviate nociception. N: no significant response, U: the research is unclear.
IL-6
The IL-6 gene is located at the p21 region of human chromosome 7 and encodes various IL-6 subtypes with molecular weights ranging from 21.5 to 28 kDa. The IL-6 receptor is a complex protein composed of two subunits: IL-6 receptor α chain (IL-6Rα) and the signal transducing subunit (gp130). IL-6Rα is the specific subunit of the IL-6 receptor, whereas gp130 is a shared signal transducing subunit that is also a common component of many other cytokine receptors. IL-6Rα is a single transmembrane protein consisting of an extracellular domain, a transmembrane domain, and an intracellular domain. IL-6 binds to IL-6Rα subunit to form a complex. Subsequently, this complex associates with the gp130 subunit on the cell membrane, forming a ternary complex. State activation of JAK/STAT pathways initiates signal transduction pathways, leading to a series of intracellular biological effects. 25
IL-6 plays a pivotal role as an inflammatory mediator in rheumatoid arthritis, and its elevated levels are closely associated with disease progression and severity. Research indicates that in rheumatoid arthritis, IL-6 production and release primarily originate from inflammatory cells, follicular dendritic cells, and follicular helper T cells, which produce significant amounts of IL-6 upon inflammation stimulation. Elevated IL-6 levels can also induce a positive feedback loop, leading to further IL-6 elevation. 26
IL-6 initially triggers pain in primary sensory neurons by enhancing the translational machinery involved in sensory neuron signal transmission. IL-6 has been shown to stimulate the production and release of key proteins involved in sensory neuronal signal transmission. Firstly, IL-6 promotes the synthesis and secretion of nerve growth factor (NGF), thereby facilitating neuronal growth and survival. Additionally, IL-6 enhances the production of neurotrophic factors (NTFs), such as brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF), which are crucial for the maintenance and regeneration of sensory neurons. Moreover, IL-6 upregulates the expression of neuronal calcium-binding proteins (NCS), which play a pivotal role in regulating calcium ion concentrations in neurons, thereby influencing neuronal excitability and signal transmission. Furthermore, IL-6 presence induces the production of other inflammatory mediators, including prostaglandins and interleukin-1β, which can modulate the excitability and transmission properties of sensory neurons.
Both in vitro and in vivo, IL-6 can induce heat hyperalgesia responses. This process is mediated through the activation of PKC via growth factor receptor-bound protein 2-associated binding protein 1/2/phosphatidylinositol 3-kinase(Gab1/2/PI3K) and subsequent modulation of TRPV1, including upregulation of the transient receptor potential vanilloid 1 (TRPV1) and TRPV2 ion channels. 27 Upon binding to its receptor, interleukin-6 (IL-6) directly activates protein kinase C (PKC), leading to the phosphorylation of downstream target proteins such as mitogen-activated protein kinase (MAPK) and protein kinase B (PKB or Akt), thereby mediating cellular signal transduction. Additionally, IL-6 indirectly activates the Gab1/2/PI3K signaling pathway upon receptor activation. Gab1/2 interacts with the activated receptor, forming a signaling complex that subsequently activates PI3K. Activated PI3K phosphorylates phosphatidylinositol bisphosphate (PIP2), generating phosphatidylinositol trisphosphate (PIP3), which in turn activates Akt protein kinase, facilitating cell survival, proliferation, and metabolic regulation. These changes result in a lowered neuronal response threshold and peripheral sensitization. Accumulating evidence underscores the critical role of IL-6 in pain induction. For instance, intra-articular injection of IL-6 in healthy rats leads to long-term sensitization of C fibers in the knee joint. 28 In an animal model of antigen-induced arthritis, intra-articular injection of soluble gp130 (an IL-6 antagonist) can alleviate pain without reducing inflammation, while systemic administration of gp130 does not yield significant effects. Rats without sensory abnormalities exhibit minimal IL-6 response. Co-administration of soluble gp130 (sgp130) can prevent IL-6-induced sensitization, but it has not been observed to reverse sensitization. 29 Importantly, these findings suggest a greater role for IL-6 during the induction phase of pain sensitization, indicating the potential efficacy of early IL-6 antagonism.
IL-1β
The IL-1 family of cytokines (IL-1F) comprises 11 proteins that share similar structures and play various roles in inflammation. Among them, IL-1β has been identified as a key inducer of inflammation and structural damage in arthritis. IL-1β contributes to pain and pain hypersensitivity. The significant upregulation of IL-1R in the dorsal root ganglia (DRGs) and periarticular tissues of the adjuvant-induced arthritis (AIA) model and collagen-induced arthritis (CIA) rats indicates the crucial involvement of IL-1 family cytokines and their receptors in these disease models. This upregulation may contribute to the exacerbation of inflammatory responses, structural damage, and pain generation. In the DRGs, increased expression of IL-1R may render sensory neurons more sensitive to pain signals, leading to the occurrence of pain hypersensitivity. 30 These findings provide a foundation for understanding the pathogenesis of rheumatoid arthritis and offer important insights for further investigating therapeutic strategies targeting IL-1 family cytokines.
In rats, intraplantar injection of IL-1β can induce cutaneous hyperalgesia and transient ongoing discharge. 31 Interestingly, IL-1β sensitizes joint C fibers to mechanical stimulation while significantly reducing the sensitivity of Aδ fibers. Since Aδ and C fibers converge on the same spinal neurons, the resulting joint pain is a manifestation of combined effects. Intraplantar injection of IL-1β increases the responsiveness of spinal dorsal horn neurons in rats. It also induces mechanical hyperalgesia and allodynia. 32 Studies have demonstrated that the presence of IL-1β in the sciatic nerve or DRG leads to direct and rapid activation of TRPV1+ DRG sensory neurons. 33 Anakinra, a biologic agent also known as recombinant human interleukin-1 receptor antagonist (rhIL-1ra), is primarily used for treating inflammatory diseases such as rheumatoid arthritis. The combined intrathecal administration of anakinra, along with soluble TNF receptors, has demonstrated pronounced analgesic properties in a rat model of neuropathic pain induced by L5 spinal nerve transection. 34 Benoit Mailhot’s study using conditional gene knockout technology specifically blocking IL-1R1 signaling in neurons revealed the essential role of neuronal IL-1R1 in mechanical pain hypersensitivity induced by RA. 35 Moreover, the incidence of IL-1-induced mechanical allodynia is lower compared to heat-induced hyperalgesia. Since RA-related pain is predominantly mechanical, IL-1 antagonists such as anakinra may not alleviate pain caused by RA. This suggests that mechanical pain hypersensitivity and thermal hyperalgesia are mediated by distinct peripheral mechanisms. Research indicates that subcutaneous injection of IL-1β produces mechanical hyperalgesia through peripheral PKA signaling and phosphorylation of peripheral NMDA/non-NMDA ion channels, leading to primary afferent fiber sensitization. 36 On the other hand, IL-1β-induced thermal hyperalgesia is mediated by peripheral PKC activation and phosphorylation of peripheral TRPV1. 20 Thus, IL-1β primarily plays an inducing role in pain sensitization, and the efficacy of its antagonists in reversing sensitization and providing pain relief is limited.
IL-17
In a recent study examining IL-17’s role in pain among rheumatoid arthritis patients and its modulation of the inflammatory response associated with neuropathic pain resulting from nerve injury, it was discovered that IL-17 plays a crucial role. In male adjuvant-induced arthritis mice, elevated IL-17 levels were observed in the joint vicinity. 37 Interestingly, mice deficient in this cytokine displayed enhanced resistance to arthritis induction in various experimental models. 38 The present study highlights the potential of IL-17A to induce long-term activation of intracellular signaling pathways and the sustained upregulation of transient receptor potential vanilloid receptor 4 (TRPV4) in DRG neurons, while TRPV1 upregulation was not observed. IL-17A-deficient mice exhibited reduced mechanical nociceptive sensitization but not thermal nociceptive sensitization, predominantly affecting C-fibers. However, at very low doses, the reduction of IL-17A led to a diminished response of Aδ fibers. In mice, intra-articular administration of IL-17 resulted in increased production of TNF-α, IL-1β, and CXCL1, leading to mechanical hypersensitivity and neutrophil recruitment. Moreover, IL-17A stimulation in isolated DRG neurons led to enhanced expression of TRPV4. 33 Notably, IL-23 indirectly induced mechanical pain in female rheumatoid arthritis patients through the release of IL-17A from T cells, innate lymphocytes and an unusual type of neutrophil, while the involvement of C-fiber injury receptors and TRPV1 was also necessary. 39 Although these findings suggest the involvement of IL-17 in mechanical nociceptive sensitization in rheumatoid arthritis patients, further in-depth investigation into IL-17 is warranted.
Granulocyte-colony-stimulating factor
Granulocyte-colony-stimulating factor (GM-CSF) is a monomeric glycoprotein composed of 174 amino acids, primarily produced by activated T cells, macrophages, and fibroblasts. 40 It exerts its effects through multiple signaling pathways, including the JAK2/STAT5 pathway, and also stimulates the release of various pro-inflammatory cytokines and chemokines, such as CCL17. The pivotal role of GM-CSF in the development of arthritis and pain has been extensively demonstrated. In an adjuvant-induced arthritis (AIA) model, mice lacking GM-CSF (GM-CSF-/-) showed significantly reduced joint swelling compared to wild-type mice following injection of mBSA. Moreover, joint destruction was markedly attenuated in GM-CSF-/- mice compared to wild-type mice 6 weeks after intra-articular injection. This reduction in joint destruction coincided with diminished mechanical nociceptive sensitization, indicating the involvement of GM-CSF in mediating mechanical sensitization responses. 41 Subsequent investigations using a murine model of bone cancer pain further elucidated the role of GM-CSF. Researchers evaluated mechanical and thermal hypersensitivity using various tests and studied the effects of hematopoietic colony-stimulating factors (HCSFs) on hypersensitivity by administering neutralizing antibodies and inhibitors. The findings revealed the significant involvement of HCSFs, including GM-CSF, in both mechanical and thermal hypersensitivity, shedding light on the mechanisms underlying tumor-nerve interactions and bone cancer pain. 42 Increased levels of GM-CS were also observed in the cytokine profiles of synovial fluid and blood samples from rheumatoid arthritis patients. Additionally, GM-CSF drives the regulatory effects of other immune cells, leading to the secretion of cytokines and the formation of pro-inflammatory cytokine networks, such as the direct induction of CCL17 and the indirect promotion of IL-1β, TNF-α, IL-6, and IL-10 release, which contribute to nociceptive sensitization. 43 Notably, GM-CSF plays a crucial role not only in rheumatoid arthritis but also in cancer pain. 6 The main mechanism by which GM-CSF induces nociceptive sensitization is the direct activation of receptors located on primary afferent nerve fibers, resulting in peripheral sensitization. Collectively, these studies underscore the significant role of GM-CSF in pain regulation.
Tumor necrosis factor-alpha
TNF-α, a pro-inflammatory cytokine, plays a crucial role in pain modulation and is implicated in various pathological conditions, including chronic inflammation and neuropathic pain. TNF-α exerts its effects by binding to its two receptors, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2), which have been identified in the dorsal root ganglia (DRG) based on animal studies. 44 The interaction of TNF-α with these receptors contributes to the development and maintenance of hyperalgesia in chronic inflammatory states. 37
In addition to its role in hyperalgesia, TNF-α has been shown to induce synaptic plasticity, a phenomenon associated with central sensitization and the amplification of pain signaling. By acting on various regions of the nervous system, including the spinal cord, thalamus, periaqueductal gray, and amygdala, TNF-α promotes synaptic modifications and enhances pain transmission. 24 These neuroplastic changes can lead to the persistence of pain even after the initial injury or inflammation has subsided, contributing to the establishment of chronic pain state.
Furthermore, TNFR1 has been implicated in mediating neuropathic pain, a type of pain resulting from damage or dysfunction of the peripheral or central nervous system. 20 Following peripheral nerve injury, there is an upregulation of TNFR1, TNFR2, and TNF-α expression, suggesting their involvement in the pathophysiology of neuropathic pain. 45 The activation of TNF-α signaling pathways in injured nerves and surrounding tissues can contribute to the development of neuropathic pain by promoting neuroinflammation, neuronal hyperexcitability, and aberrant sensory processing. Importantly, targeting TNF-α has emerged as a therapeutic strategy for managing chronic pain. Reduction of TNF-α levels, either through pharmacological interventions or genetic manipulations, has been shown to reverse the altered connectivity in the upper circuitry of the central nervous system, leading to a reduction in chronic pain state. 46 Clinical studies have demonstrated the efficacy of TNF-α inhibitors in alleviating pain and improving functional outcomes in patients with conditions such as rheumatoid arthritis and neuropathic pain.
Therapeutic targeting of the JAK/STAT pathway
JAK inhibitors, also known as JAK kinase inhibitors or JAKi, are a class of therapeutic agents that specifically target the Janus kinase family of enzymes. JAK inhibitors exert their pharmacological effects by selectively inhibiting the activity of JAK enzymes, thereby modulating the signaling pathways associated with cytokines and growth factors. 47 JAK inhibitors have shown significant potential in the treatment of various inflammatory and autoimmune diseases, such as rheumatoid arthritis, especially for patients with inadequate response levels to traditional DMARDs. However, the risk of concomitant infection and venous thrombosis is also greater. 48 JAK inhibitors offer a new option in rheumatoid arthritis, and their widespread use stems from the close association of the JAK/STAT pathway with the pathogenesis of rheumatoid arthritis. The precise targeting of these biologics has resulted in better control of disease activity and progression of joint damage in rheumatoid arthritis patients compared to traditional DMARDs. Tofacitinib and baricitinib, as first-generation pan-JAK inhibitors, have the function of blocking a variety of cytokines and are widely used because of their safety and efficacy.
Tofacitinib is a potent oral small molecule JAK inhibitor that acts primarily targeting JAK1 and JAK3, with lower activity for JAK2 and TYK2, and is an innovative advance in the treatment of rheumatoid arthritis. The double-blind, parallel, placebo-controlled study conducted by Ronald F et al. demonstrates that both doses of tofacitinib (5mg and 10mg, BID) effectively reduce pain in many patients, as supported by relevant clinical trials. 49
Baricitinib is a recently marketed jak inhibitor that acts primarily against the JAK1 and JAK2 pathways, with moderate activity for TYK2. It is approved for use as monotherapy or in combination with methotrexate for the treatment of adult patients with moderately severe active rheumatoid arthritis who have had an inadequate response or are intolerant to more than one DMARD. A Randomized, Double-blind, Multicenter, Phase 3 Study of the Efficacy and Safety of Baricitinib in Patients with rheumatoid arthritis(RA-BEAM) is a clinical trial aimed at evaluating the effectiveness and safety of Baricitinib, a medication used in the treatment of rheumatoid arthritis. 50 The trial compared disease activity, and joint functional impairment in the baricitinib, adalimumab and placebo groups, where pain was measured on a visual analog scale (VAS) 0–100 mm assessment. The results showed superior efficacy of baricitinib monotherapy over methotrexate monotherapy at 24 weeks (p < .01). There was a statistically significant improvement in the efficiency of baricitinib monotherapy and baricitinib + MTX compared to MTX monotherapy for ACR20, ACR50 and ACR70. Not only that, compared to adalimumab and placebo, baricitinib had a more rapid clinical pain relief effect. Specifically, the time required to achieve ≥50% pain relief in the baricitinib group was half the time required for adalimumab. Rheumatoid arthritis is commonly associated with residual pain during treatment, which is typically assessed using visual analog scale (VAS) or numeric rating scale (NRS). It is widely acknowledged that residual pain ≤20 mm does not significantly impact patient satisfaction with health outcomes. In comparison to placebo + MTX, the combination of Baricitinib and MTX is more feasible and likely to achieve pain levels of ≤20 mm. Additionally, patients treated with Baricitinib exhibit greater improvement in pain at 6 months compared to those treated with adalimumab and tocilizumab. Thus, these findings suggest that Baricitinib in combination with MTX holds promising potential in reducing pain in RA patients. 51
In clinical trials, it has been observed that baricitinib treatment significantly reduces symptoms of arthritis, including joint pain, morning stiffness, and joint swelling, while improving joint function and mobility in patients. Furthermore, baricitinib has shown potential in improving fatigue and sleep quality. In comparison to traditional disease-modifying drugs, baricitinib exhibits faster onset of action and more effective modulation of arthritis-induced pain sensitization. Although some patients may experience mild adverse reactions such as headache, gastrointestinal discomfort, and mild immunosuppression, overall, baricitinib demonstrates remarkable efficacy in alleviating pain symptoms and reducing inflammation levels. These clinical observations indicate that baricitinib is a promising therapeutic option that can significantly alleviate pain and related symptoms in patients with chronic inflammatory arthritis. However, further long-term research and clinical practice are necessary to determine its sustained efficacy and safety profile. 52
In addition, some biological agents can also improve the quality of life of patients by reducing hyperalgesia indirectly through the JAK/STAT pathway. Adalimumab, a humanized monoclonal antibody, exerts its effects on tumor necrosis factor-alpha (TNF-α). TNF-α plays a crucial role in the inflammatory process and is considered a key factor in pain and hyperalgesia in patients with rheumatoid arthritis (RA). Adalimumab binds and neutralizes TNF-α, thereby exerting anti-inflammatory and immunomodulatory effects in rheumatoid arthritis. Numerous studies have demonstrated the significant impact of adalimumab in reducing pain sensitization. Firstly, its anti-inflammatory properties alleviate the release of inflammatory mediators and dampen the inflammatory response, thereby reducing joint tissue damage and pain perception. Secondly, adalimumab disrupts the interaction between inflammatory cells and neurons, inhibiting the transmission of inflammatory signals and decreasing pain signal generation and propagation. Furthermore, adalimumab inhibits neuronal excitability and synaptic plasticity, further contributing to the reduction of pain sensitization. By suppressing the direct effects of inflammatory mediators on neurons, adalimumab mitigates neuronal hyperexcitability and aberrant discharges, thereby alleviating pain sensation. 51
Tocilizumab, a monoclonal antibody targeting the interleukin-6 receptor, has demonstrated efficacy in reducing pain sensitization and improving clinical outcomes. IL-6, a pro-inflammatory cytokine, plays a crucial role in the development and maintenance of chronic pain conditions. Tocilizumab acts by specifically binding to IL-6 receptors, thereby blocking IL-6 signaling and mitigating its effects on pain sensitization. One of the primary mechanisms by which tocilizumab reduces pain sensitization is through its potent anti-inflammatory properties. By inhibiting IL-6 signaling, tocilizumab effectively suppresses the production of various pro-inflammatory mediators, including cytokines and chemokines. This anti-inflammatory action results in the attenuation of tissue inflammation, reducing peripheral sensitization and subsequent pain perception. Tocilizumab also modulates synaptic plasticity and neuronal excitability, contributing to its analgesic effects. IL-6 signaling pathways, particularly those involving Janus kinases and signal transducer and activator of transcription proteins, are disrupted by tocilizumab. This disruption affects neuronal activity and synaptic plasticity, leading to a reduction in central sensitization and the modulation of pain responses. Clinical studies have shown that tocilizumab treatment leads to a significant improvement in pain symptoms and functional outcomes in patients with conditions such as rheumatoid arthritis. Patients receiving tocilizumab experience reduced joint tenderness, swelling, and morning stiffness, along with improvements in physical function and quality of life. Furthermore, tocilizumab treatment has been associated with a decrease in the use of pain medications, highlighting its efficacy in pain management. It is worth noting that tocilizumab’s clinical effects on pain sensitization extend beyond its direct analgesic properties. By targeting IL-6, tocilizumab modulates the immune response, influencing the activity of immune cells involved in pain modulation, such as macrophages and T cells. This immunomodulatory effect contributes to the overall reduction of pain sensitization and the improvement of pain-related symptoms. 53
Hence, these collective findings provide robust evidence supporting the therapeutic efficacy of JAK inhibitors and biologics indirectly acting through JAK/STAT pathway, which exert their pain-relieving effects in rheumatoid arthritis. Importantly, these analgesic properties align with the well-established role of the JAK pathway in mediating the pro-inflammatory regulatory actions of various cytokines.
Conclusions
Pain management is a crucial component in the treatment of rheumatoid arthritis, and the impact of pain on rheumatoid arthritis patients should not be underestimated. The etiology of pain in rheumatoid arthritis patients is multifaceted, involving both inflammatory and non-inflammatory factors, with the latter encompassing peripheral sensitization and central sensitization. Both peripheral sensitization and central sensitization contribute to the modulation of nociceptive receptors. Throughout the development of chronic synovitis, immune cells within the joint capsule, alongside resident cells, modulate nociceptive receptors by releasing cytokines, thereby reducing the receptor threshold and inducing sensitization. Many of these cytokines engage in intracellular signaling via the JAK/STAT pathway, which in turn regulates pain perception. Consequently, the pain-alleviating effects of Janus kinase inhibitors align with their demonstrated clinical efficacy. Nevertheless, further investigations are necessary to elucidate whether inhibition of a singular signaling pathway can holistically improve pain outcomes in rheumatoid arthritis patients.
Footnotes
Acknowledgements
We thank participants at the Conferences you have attended for valuable discussions and comments.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Natural Science Foundation of China (82101887), the Suzhou Minsheng science and technology project (SYS2019043 and SYS2020108), and Beijing Health Alliance Charitable Foundation (B21046ES).