
Abstract
Her2 is a receptor tyrosine kinase overexpressed in 25% of breast tumors. We have shown that the 88 kDa autocrine growth and survival factor GP88 (progranulin) stimulated Her2 phosphorylation and proliferation and conferred Herceptin resistance in Her2-overexpressing cells. Herein, we report that GP88 stimulates c-myc phosphorylation and upregulates c-myc levels in Her2-overexpressing cells. c-myc phosphorylation and upregulation by GP88 were not observed in non-Her2-overexpressing breast cancer cells. c-myc activation was inhibited upon treatment with ERK, PI3 kinase, and c-src pathway inhibitors, U0126, LY294002, and PP2. GP88 also stimulated c-src phosphorylation, a known upstream regulator of c-myc. Thus, we describe here a signaling pathway for GP88 in Her2-overexpressing cells, with GP88 stimulating Src phosphorylation, followed by phosphorylation and upregulation of c-myc. These data would suggest that targeting GP88 could provide a novel treatment approach in breast cancer.
Introduction
The erbB family of receptor tyrosine kinases plays a critical role in cancer. This family consists of four members, namely, erbB1/EGFR, erbB2/Her2, erbB3/Her3, and erbB4/Her4.1,2 Except for erbB2/Her2 that lacks an accessible ligand-binding domain, 3 each member of the erbB receptor family has several ligands. Upon ligand binding, these receptors undergo either homodimerization or heterodimerization, leading to transphosphorylation and initiation of signaling cascades, including both MAP kinase and PI3 kinase pathways, leading to cell proliferation and survival.4,5 Of all the possible pairings between the family members, the erbB2/erbB3 heterodimers are the most abundant and potent signaling modules formed.6,7 ErbB2/Her2 is overexpressed in numerous human cancers such as breast, ovarian, gastric, colon, and non-small cell lung cancers.8,9 Overexpression of erbB2/Her2 in breast cancer is found in about 25% of breast carcinoma and is associated with poor prognosis. 10 Cells overexpressing erbB2/Her2 are more invasive and resistant to chemotherapy and endocrine therapy.11,12 The most common treatment for patients with breast tumors overexpressing erbB2/Her2 is the use of the humanized monoclonal antibody trastuzumab, also known as Herceptin®. Herceptin binds to the extracellular domain of the Her2 receptor, and this binding is believed to cause homodimerization, a pairing that is ineffective in the activation of oncogenic downstream signaling, resulting in Her2 downmodulation. 13 Treatment with Herceptin causes a cytostatic growth inhibitory effect in breast cancer cells overexpressing Her2.14––16 As a single agent, Herceptin is effective in approximately 20% of the treated patients. 17 In combination therapy with chemotherapeutic agents such as taxotere, the response rate is increased to 50%. 18 Recently, a second generation of anti-Her2 antibody called pertuzumab was approved by the Food and Drug Administration for use in combination with Herceptin and docetaxel. Pertuzumab (Perjeta) prevents the heterodimerization of Her2 with other members of the Her family, particularly Her-3. 19 It is thought that Perjeta efficacy will be determined by addressing patients having lower Her2 expression or patients showing resistance to Herceptin. 20 Several mechanisms of Herceptin resistance have been described. They include hyperactivation of the phosphatidylinositol-3-kinase (PI3K) pathway, coexpression of the truncated p95Her2 receptor, heterodimerization with other growth factor receptors, loss of Her2 expression, and upregulation of signaling molecules.21,22
Our laboratory has identified the 88 KDa glycoprotein autocrine growth/survival factor GP88 (also known as progranulin, acrogranin, granulin/epithelin precursor, or PC cell-derived growth factor) as a biological driver of tumorigenesis in several cancers, including breast cancer. 23 GP88 expression is associated with increased proliferation and survival, leading to resistance to current breast cancer therapies such as doxorubicin, Herceptin, and antiestrogens. 23 GP88/progranulin is the largest member of a unique family of cysteine-rich polypeptides that encompasses seven and a half 6 kDa epithelin or granulin repeats into a 63 kDa core protein with a 17 amino acid signal peptide targeting GP88 for secretion.24,25 For several cancer types, GP88 has been shown to be involved in proliferation, survival, migration, angiogenesis, invasion, and matrix metalloprotease activity. 26 In addition, in normal tissues, it plays a role in wound healing, inflammation, and neuronal development.27,28 The pathways involved in GP88 signaling include both the mitogen-activated protein kinase (MAP kinase ERK1/2), phosphatidylinositol-3-kinase (PI3 kinase), and focal adhesion kinase, leading to the activation of the cell cycle regulatory proteins Cyclin D1 and Cyclin B.29,30 In ER-positive (ER+) breast cancer cells, GP88 expression was associated with the acquisition of resistance to antiestrogen tamoxifen, aromatase inhibitor, and Faslodex.31––34
Immunohistochemistry (IHC) studies of formalin-fixed paraffin-embedded tumor specimens using the anti-human GP88 antibody 6B3 developed in our laboratory have shown that GP88 expression is low or negative in normal mammary tissues but elevated in the corresponding malignant tissues. 35 Recent training and validation studies totaling 600 estrogen receptor-positive invasive ductal carcinoma (ER+ IDC) cases established a GP88 IHC cutoff score associated with poor prognosis and demonstrated that high GP88 IHC expression (score 3+) was statistically associated with a 5.9-fold higher hazard of disease recurrence and a 2.5-fold higher mortality hazard compared with patients with tumor GP88 IHC score <3+. GP88 expression remained an independent risk predictor after considering age, ethnicity, nodal status, tumor size, tumor grade, disease stage, progesterone receptor expression, and treatments. 36 Additionally, a GP88 enzyme immunoassay developed in our laboratory to measure circulating levels of GP88 showed in a prospective longitudinal clinical study that serum GP88 levels in breast cancer patients were elevated when compared with serum GP88 level from healthy volunteers. 37 Pathological studies described above also indicated that high GP88 (score 3+) was found in 25% of Her-overexpressing IDC. 35 Biological studies performed with Her2-overexpressing breast cancer cells lines such as MCF-7/Her2, SKBR3, and BT474 showed that GP88 stimulated Her2 phosphorylation in a time- and dose-dependent fashion and conferred resistance to the anti-Her2 receptor antibody Herceptin and thus must activate signaling pathways downstream of Her2. 38
One interesting biomarker upregulated in breast cancer, found downstream of Her2 and associated with Her2 therapy resistance, is c-myc. 39 In fact, c-myc is a primary effector of Her2-mediated oncogenicity. 40 c-myc is a transcription factor known to modulate both cellular proliferation and apoptosis under specific growth conditions in breast cancer.41––43 c-myc is also a downstream target of the p42/44 MAP kinase. 43 Based on the above findings and since GP88 is associated with Herceptin resistance, we investigated whether GP88 could activate c-myc. The present study shows that GP88 not only increases the levels of c-myc but also activates c-myc in a time-dependent fashion in Her2-overexpressing cells.
Materials and Methods
Cell lines and antibodies
SKBR3 cells and MCF-7 cells were obtained from ATCC. MCF-7 cells and stable transfectants derived from these cells were cultivated in DMEM/F12 medium (1:1) supplemented with 5% fetal bovine serum (FBS; Life Technologies). Her2-overexpressing MCF-7 (MCF-7/Her2) cells were developed in our laboratory as described previously 38 and were cultivated in the presence of 500 μg/mL G418 (RPI). SKBR3 cells were cultivated in McCoy's 5A Medium (ATCC) supplemented with 10% FBS. For immunoblots, the following antibodies were used: c-myc, phospho-c-myc (Thr58/Ser62), phospho-src, (Tyr416) from Cell Signaling Technology, and β-actin (Sigma Chemical Co.). Inhibitors PP2, LY294002, and U0126 were from Calbiochem. Human recombinant GP88/progranulin was produced and purified in our laboratory at A&G Pharmaceutical Inc., as described previously.31,38
Stable transfection of HER2 cDNAs in MCF-7 cells
pcDNA3 (Invitrogen) expression vector containing the Her2 cDNA 5 (kindly provided by Dr. Yosef Yarden, Weizmann Institute of Science, Rehovot, Israel) was stably transfected into MCF-7 breast carcinoma cells using Lipofectamine Plus (Life Technologies) according to the manufacturer's protocol. Transfected cells were selected with 1.2 mg/mL G418, as described previously. 38 HER2 expression in these cells was checked by Western blot analysis using anti-Her2 antibody (Millipore).
Western blot analysis of phosphorylated c-myc and phosphorylated c-src expression
A total of 2 × 10 5 cells were plated on 35 mm2 dishes in the appropriate culture medium, according to the cell type as described above. SKBR3 cells were plated in McCoy's 5A Medium supplemented with 10% FBS. MCF-7 cells and derivatives were plated and cultured in DMEM/F12 medium supplemented with 5% FBS for 48 hours. Prior to starting the experiment, the cells were washed with serum-free phenol red free DME-F12 medium (PFMEM) medium and incubated in serum-free medium for 24 hours, and the indicated treatments were added 1 hour later. Specific inhibitors were preincubated as described below. After washing with cold phosphate buffered saline (PBS) twice, cell lysates were collected in cell lysis buffer (50 mM Tris, pH 7.4, 4 mM ethylenediaminetetraacetic acid (EDTA), 25 mM KCl, 1 mM Na3VO4, 10 mM NaF, 1% Triton 100, 10 μg/mL leupeptin, 10 μg/mL pepstatin, 2 μg/mL aprotinin, and 1 mM phenylmethylsulfonyl fluoride [PMSF]). A total of 40 μg of total protein lysates were loaded onto SDS-PAGE gels, and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% milk in PBS-T (Tween 20 0.5%) for 2 hours at room temperature. Membranes were incubated with the primary antibody overnight at 4°C. After washing with PBS-T three times, five minutes each, membranes were incubated with the corresponding secondary antibody for two hours at room temperature. Membranes were washed three times, 15 minutes each. Proteins were detected with enhanced chemiluminescence.
Results
GP88 stimulates c-Src phosphorylation in Her2-overexpressing cells
Phosphorylation of the tyrosine residues located at the carboxyl-terminal domain of Her2 provides a docking site for downstream signaling molecules, such as Src. 44 Since GP88 activates Her2 and stimulates its phosphorylation, 38 we investigated whether GP88 could stimulate c-Src phosphorylation in Her2-overexpressing cells. After treating MCF-7/Her2 cells with 300 ng/mL of pure human GP88 for three minutes, we observed an increase in c-Src phosphorylation (Fig. 1A).

GP88 activates Src in Her2-overexpressing cells MCF-7/Her2, Her2 transiently transfected Cos-7 cells, and SKBR3 cells. (A) 2 × 10 5 MCF-7/Her2 cells were plated on 35 mm dishes. Forty-eight hours later, cells were washed, and fresh PFMEM was added, and 24 hours later, either 300 ng/mL GP88 (G) or 100 ng/mL EGF (E) was added. Control cells (C) received an equivalent volume of PBS. Thirty minutes prior to adding growth factors, cells were treated either with PP2 (100 nM) (+) or with vehicle control (–). Cell lysates were collected three minutes later. Western blot analysis was performed with anti-phospho-Src antibody (Cell Signaling Technology). Membranes were reprobed with anti-actin antibody to ensure equal loading. (B) Cos-7 cells were transfected with pcDNA3/Her2 construct using Lipofectamine. Twenty-four hours after transfection, cells were washed and fresh PFMEM was added, and 24 hours later, either vehicle (C), 300 ng/mL GP88 (G) or 100 ng/mL EGF was added (E). PP2 (100 nM) was added (+) or not (–) 30 minutes prior to adding GP88. Cell lysates were collected three minutes later. Western blot analysis was performed with anti-phospho-Src antibody (Cell SignalingTechnology). Membranes were reprobed with anti-actin antibody to ensure equal loading. (C) 2 × 10 5 SKBR3 cells were plated in McCoy's 5A Medium supplemented with 10% FBS in 35 mm dishes. Forty-eight hours later, cells were washed, and fresh PFMEM was added, and 24 hours later, either 300 ng/mL GP88 (G) or 100 ng/mL EGF (E) was added. Control cells (C) received an equivalent volume of PBS. Cells were collected five minutes later for measuring p-src activation as described above.
To rule out the possibility that Src was being exclusively activated through the estrogen receptors present on MCF-7 cells, 45 Cos-7 cells were transiently transfected with Her2 cDNA and treated with GP88. We found that GP88 stimulated c-Src phosphorylation in Her2-overexpressing Cos-7 cells in a similar fashion as in MCF-7/Her2 cells (Fig. 1B). These results suggested that GP88 rapidly phosphorylated c-Src through Her2 activation in Her2-overexpressing cells.
Interestingly, PP2, an Src kinase inhibitor, could not inhibit the levels of c-Src phosphorylation in the presence of GP88 (Fig. 1A and B). In contrast, EGF's ability to phosphorylate c-Src in the same cells was decreased by PP2, as expected. This suggests that the activation of c-Src upon GP88 treatment is not necessarily the result of direct Src kinase activation and Src autophosphorylation, but rather perhaps through an interaction with Her2. Since we previously demonstrated that GP88 phosphorylates Her2, 38 and Src binds to the C-terminal domain of Her2, the observed phosphorylation of Src could be the immediate, subsequent step following the GP88-induced Her2 activation.
GP88 was also able to stimulate phosphorylation of c-Src in a dose-dependent fashion (Fig. 1C) in SKBR3 cells, another well-studied breast cancer cells naturally overexpressing Her2, indicating that GP88 effect was not restricted to Her2-transfected cells.
GP88 stimulates phosphorylation of c-myc in Her2-overexpressing cells
We previously demonstrated that GP88 activates ERK1/2 in Her2-overexpressing breast cancer cells. 38 Since c-myc is a downstream target of Her2 and ERK1/2, we examined whether GP88 could activate c-myc in MCF-7/Her2 cells. The cells were treated with GP88 in a time-dependent fashion. Phosphorylation of c-myc was observed and transiently peaked at 15 minutes following GP88 addition similar to the Her2 activator Heregulin, used as positive control. Phosphorylation of c-myc returned to basal level 30 minutes after GP88 addition (Fig. 2A).

Effect of GP88 on c-myc phosphorylation in Her2-overexpressing or Her2-nonexpressing breast cancer cells. (A): MCF-7/Her2 cells were treated with 300 ng/mL GP88 (G), as described in the “Materials and methods” section. As positive control, cells were treated with 5 ng/mL Heregulin β1. Cells were collected at the indicated time points to prepare cell lysates. A total of 40 μg of total protein was resolved on a 7.5% SDS polyacrylamide gel. Western blot analysis was performed with anti-phospho-c-myc. Actin expression was measured as internal control for equal loading. Membranes were reprobed with anti-actin to ensure equal loading. (B) MCF-7 cells were treated with 300 ng/mL of purified GP88 or 5 ng/mL Heregulin β1 as described in the “Materials and methods” section. Cell lysates were prepared at the indicated time points. A total of 40 μg of total protein was resolved on a 7.5% SDS polyacrylamide gel. Western blot analysis was performed as described in (A). C, control; G, GP88; H, Heregulin β1. (C): 2× 10 5 SKBR3 cells were plated in McCoy's 5A Medium supplemented with 10% FBS in 35 mm dishes. Forty-eight hours later, cells were washed, and fresh PFMEM was added, and 24 hours later, cells were treated for 5 minutes with either 300 ng/mL GP88 (G) or 5 ng/mL Heregulin (H). Control cells (C) received an equivalent volume of PBS. Western blot analysis was performed as described in (A).
We have shown previously that GP88 also stimulates the proliferation of non-Her2-overexpressing cells such as MCF-7 cells. 31 Based on this result, we examined whether GP88 could stimulate c-myc phosphorylation in MCF-7 cells. Interestingly, when MCF-7 cells that do not overexpress Her2 were treated with GP88, no phosphorylation of c-myc was observed in contrast to EGF that shows a robust and sustained stimulation of c-myc phosphorylation (Fig. 2B). These results show that Her2 overexpression is facilitating GP88's ability to stimulate c-myc phosphorylation. Stimulation of c-myc phosphorylation by GP88 was also observed in Her2-overexpressing breast cancer cell line SKBR3 (Fig. 2C).
GP88 stimulates c-myc phosphorylation concomitantly through ERK1/2 and PI3-kinase
Her2 utilizes both the ERK1/2 and PI3 kinase signaling pathways. Therefore, we investigated whether the ability of GP88 to stimulate c-myc phosphorylation was via either the ERK1/2 or PI3 kinase pathways. We treated MCF-7/HER2 cells with GP88 in the presence of specific ERK1/2 and PI3 kinase inhibitors, U0126 and LY294002, respectively. We found that the phosphorylation of c-myc by GP88 was reduced in the presence of either of these inhibitors (Fig. 3A). These findings confirm that the activation of c-myc by GP88 is dependent on both ERK1/2 and PI3 kinase.
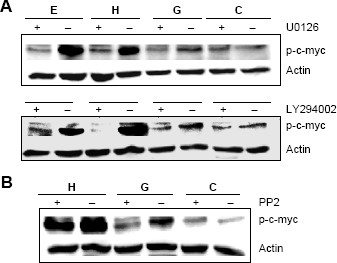
Effect of pathway inhibitors on the ability of GP88 to stimulate c-myc phosphorylation. 2 × 10 5 MCF-7/erbB2 cells were as described in the “Materials and methods” section. Twenty-four hours later, fresh PFMEM was added. (A, top panel): U0126 (20 μM) (A, bottom panel) LY294002 (10–5 M) or (B) PP2 (100 nM) were added (+) or not (–) 30 minutes prior to the addition of 300 ng/mL GP88, 10 ng/mL Heregulin β1, or 100 ng/mL EGF. Cell lysates were collected 15 minutes later. Western blot analysis was performed with anti-phospho-c-myc antibody, and membranes were reprobed with anti-actin antibody.
Src kinase inhibitor, PP2, inhibits GP88's ability to phosphorylate c-myc
As seen in Figure 1, we demonstrated that GP88 induced c-Src phosphorylation. However, this activation could not be inhibited by PP2, an Src kinase-family inhibitor. Here we examined the effect of PP2 on the phosphorylation level of c-myc by GP88. We observed that PP2 attenuated c-myc phosphorylation levels upon GP88 treatment (Fig. 3B), similar to the U0126 and LY294002 inhibition levels of c-myc phosphorylation shown earlier. This suggests that Src phosphorylation is indeed an early, upstream signaling event, ultimately targeting c-myc upon GP88 treatment.
GP88 stimulates c-myc expression in Her2-overexpressing breast cancer cells
In addition to stimulating phosphorylation of c-myc, previous studies from our laboratory have shown that GP88 cannot induce an increase in the levels of c-myc in MCF-7 cells (unpublished). However, when SKBR3 cells were treated with GP88, an increase in the levels of c-myc was observed (Fig. 4).

PC cell-derived growth factor (PCDGF) increases the levels of c-Myc in SKBR3 cells. 2 × 10 5 SKBR3 cells were plated on 35 mm dishes in McCoy's 5A Medium supplemented with 10% FBS. Forty-eight hours later, cells were washed and fresh PFMEM (no serum) was added, and 24 hours later, either 300 ng/mL of purified GP88 (G) or 100 ng/mL EGF (E) was added while control untreated cells received vehicle only. Cell lysates were collected at the indicated time points. A total of 40 μg of total protein was resolved on a 7.5% SDS-polyacrylamide gel. Western blot analysis was performed with anti-actin antibody. Membranes were reprobed with anti-actin to ensure equal loading.
This would indicate that GP88 has the ability to modulate c-Myc level as well as its activation level.
Discussion
GP88 (progranulin) is an autocrine growth and survival factor, the overexpression of which is associated with poor prognosis in early-stage breast cancer patients. 36 In Her2-overexpressing cell lines, we have previously established that GP88 stimulates Her2 phosphorylation in a time- and dose-dependent fashion followed by activation of ERK1/2 and Akt pathways. To determine the complete signal transduction pathway utilized by GP88 in Her2-overexpressing cells, the present study investigated the signaling molecules involved in stimulating Her2-mediated proliferation by GP88.
Using MCF-7/Her2 cells and SKBR3 cells, we show here that the signaling cascade triggered by GP88 upon Her2 activation includes the phosphorylation of Src, leading to the stimulation of intermediate molecules (ERK1/2 and Akt), and ultimately stimulating the transcription factor c-myc phosphorylation. In addition, GP88 upregulated the level of c-myc expression in these cells.
GP88 phosphorylated Src, an adaptor protein which is bound to the cytoplasmic domain of Her2.44––46 The fact that PP2, an Src kinase inhibitor, could not attenuate the levels of Src phosphorylation upon GP88 treatment would suggest that this phosphorylation was not due to Src autophosphorylation but most likely via Her2. Recent studies have shown that Her2 activates and upregulates Src, and this is required for Her2-mediated breast cancer metastasis 46 and drug resistance. 47 Another possibility is that the GP88 receptor can directly interact and phosphorylate Src as well. However, since GP88 cannot stimulate c-myc phosphorylation in the absence of Her2-overexpression, this would suggest that Src is initially binding to Her2, rather than to the GP88 receptor in this particular signaling paradigm. However, there is a possibility that Src could indeed bind to the cytoplasmic region of the GP88 receptor in a completely separate signaling pathway, which may even result in a cross-talk between the two pathways. It would then be assumed that GP88 binds to its receptor, and then the receptor forms a heterodimer with Her2 to initiate a proliferation signal. Studies demonstrating that Her2 is able to heterodimerize with receptors outside of its family, such as IGF-1R and estrogen receptor,48,49 would support this hypothesis. Whether Her2 and GP88 receptors heterodimerize will remain unsolved until the GP88 receptor on breast cancer cells has been completely characterized and this possibility directly investigated. However, we have evidence suggesting that GP88 cannot activate Her2 without the presence of its receptor (unpublished data). Even though two possible candidates for progranulin receptors have been identified,50,51 their definite proof of their function as progranulin receptors remain controversial52,53 and neither one has been proven to be the signaling GP88 receptor in cancer cells.
Another interesting finding from our studies was the ability of GP88 to upregulate the protein levels of c-myc, in addition to inducing its phosphorylation as well, in MCF-7/erbB2-overexpressing breast cancer cells. The ability of GP88 to stimulate c-myc phosphorylation in Her2-overexpressing cells, but not in cells with low levels of Her2 expression represents a novel signaling pathway for GP88. Since c-myc is a downstream target of MAPK/ERK, and GP88 can activate MAPK/ERK in cells that do not overexpress Her2,31,38 this suggests a GP88 signaling pathway divergence downstream of MAPK/ERK.
To further investigate and delineate this novel signaling mechanism, we investigated the ability of specific inhibitors to attenuate the levels of c-myc phosphorylation induced by GP88. Both MEK and PI3 kinase inhibitors (U0126 and LY294002, respectively) were able to decrease the levels of c-myc phosphorylation upon GP88 treatment, implying that c-myc is a common target shared by both pathways. Furthermore, the fact that LY294002 inhibited GP88's ability to phosphorylate c-myc suggests an involvement of the PI3 kinase/Akt signaling pathway. This was somewhat unexpected since in Her2-overexpressing cells, GP88 utilizes primarily the MAPK signaling pathway for inducing cell proliferation,24,31 and conferring Herceptin resistance. 38 However, previous studies have shown that Heregulin β1 increases the c-myc protein levels in Her2-overexpressing cells through the PI3K/Akt/mTOR pathway. 43 Therefore, this demonstrates the diverse signaling ability of GP88 in Her2-overexpressing cells, which could possibly explain its highly tumorigenic and therapy-resistant properties.
Furthermore, we demonstrated that PP2 attenuates the levels of c-myc phosphorylation induced by GP88. This further strengthens our proposed paradigm that Src activation is an early event in GP88-mediated signaling in erbB2-overexpressing breast cancer cells, ultimately leading to the phosphorylation of c-myc. A further, comprehensive delineation of this particular signaling pathway is necessary and could thus prove to be integral in developing novel approaches to targeted therapy.
Our previous studies have shown that GP88 confers Herceptin resistance and stimulates proliferation-associated signaling pathways in Her2-overexpressing breast cancer cells. 38 We hypothesize that the signaling mechanism described above that involves c-myc phosphorylation may be involved at least in part for the Herceptin resistance and growth advantage observed when GP88 is overexpressed in Her2-overexpressing breast cancer cells. 38 In fact, c-myc overexpression has been associated with poor prognosis in breast cancer cells, although association with Herceptin resistance remains controversial. 54
Based on our results, we hypothesize that cotargeting GP88 and Her2 may lead to a higher response rate compared with patients receiving Herceptin alone. Previous studies from our laboratory have shown that using neutralizing antibodies and antisense oligonucleotides that targeted GP88 resulted in inhibition of tumorigenicity. Since GP88 is strongly overexpressed in approximately 20% of breast carcinomas, blocking the expression or action of GP88 may represent a beneficial therapeutic opportunity. Furthermore, these findings not only reaffirm the necessity for targeting GP88 but also reaffirm the therapeutic potential in developing drugs that target Src or c-myc.
Footnotes
Acknowledgments
The authors wish to thank Dr Yosef Yarden (Weizmann Institute of Science, Rehovot, Israel) for providing the erbB2 cDNA construct, Dr. Changsheng Tian for suggestions during the transfection experiments, Dr. Paul Shapiro (University of Maryland, Baltimore), and Dr. Jun Hayashi for critical discussion and review of the manuscript and David Hicks for his help in preparing the manuscript.
Author Contributions
Conceived the study: GS. Designed the experiments: WEK and GS. Carried out the experiments: WEK and BY. Analyzed the data: WEK, BY, and GS. Wrote the first draft of manuscript: WEK. Wrote and finalized the manuscript: WEK and GS. Agreed on results and approved presentation of data in final manuscript: WEK, BY, and GS. All authors reviewed and approved of the final manuscript.