
Abstract
Two-component systems (TCS) are short signalling pathways generally occurring in prokaryotes. They frequently regulate prokaryotic stimulus responses and thus are also of interest for engineering in biotechnology and synthetic biology. The aim of this study is to better understand and describe rewiring of TCS while investigating different evolutionary scenarios.
Based on large-scale screens of TCS in different organisms, this study gives detailed data, concrete alignments, and structure analysis on three general modification scenarios, where TCS were rewired for new responses and functions: (i) exchanges in the sequence within single TCS domains, (ii) exchange of whole TCS domains; (iii) addition of new components modulating TCS function.
As a result, the replacement of stimulus and promotor cassettes to rewire TCS is well defined exploiting the alignments given here. The diverged TCS examples are non-trivial and the design is challenging. Designed connector proteins may also be useful to modify TCS in selected cases.
Keywords
Introduction
A key mechanism used by bacteria for sensing their environment is based on two-component systems (TCS). These systems typically consist of a sensor protein with a membrane-bound histidine kinase domain (HisKA) and a corresponding regulator protein with a response regulator domain (RR). The sensor protein detects specific changes in the environment and subsequently binds adenosine triphosphate (ATP). This causes a structural change of the sensor protein and, after autophorphorylation at a histidine residue, evokes phosphor-transfer to the corresponding response regulator. The response regulator then changes its structure and mediates a cellular response. 1 TCS standard structure is well conserved.2,3 Several databases describe different aspects of TCS.4–7 Mutational analyses of individual components in TCS are described in previous reports.8,9 Design, rewiring, and modifications of TCS have been studied for a long time, including efforts in biotechnology.10–16 Still, it is a major challenge to successfully engineer TCS systems, as direct design attempts only work well for controlled cases and evolutionarily short distances. 17 In taking a closer look, it turned out that information for specific cases on individual functional sites and sequences is often lacking. Therefore, we looked closely at evolutionary changes in TCS, in order to create a more solid basis for future design attempts. In synthetic biology, rewiring TCS allows us to construct synthetic networks. 18 For this, exchange of TCS promotors, partial or full replacement of sensor and regulator, as well as adding additional components is key. 19 The specific motifs involved and the overall topology of the system determine the observed switching behavior. 20
Consequently, the aim of this study is to describe and review evolutionary scenarios as a guide to rewire two-component systems.
Taking a large-scale screen on available TCS from various databases as our basis (see Supplementary material), we considered three general scenarios spanning from local to more global changes of TCS: (i) Individual amino acid changes. These lead to direct sequence changes of sensors and regulators, eg, changing specificity of stimulus or allowing the regulation of new genes. (ii) An alternative scenario considers more radical changes such as domain swapping. We performed large-scale screens and identified events in which such exchanges lead to a change in the overall function of a TCS. This can be exploited for more drastic engineering strategies, which are otherwise very difficult to predict in their outcome. (iii) Another modification strategy does not interfere with the sensor or regulator of the TCS. Additional proteins or domains, so called connectors, interact with either one or both of them. This again modulates output and performance of the TCS. Starting from a known event (SafA in Escherichia coli) we consider further proteins, which could have such connector functions and examine their potential to change TCS function.
Results and Discussion
We screened various databases for TCS and their modifications. Supplementary material illustrates this in Table S1 for a screen listing the most frequently occurring contexts in which histidine kinase or response regulator domains were found. Databases we screened include amongst others the database of protein families PFAM, 21 the protein database Uniprot, 22 as well as further repositories, such as MIST2, 4 SENTRA, 6 and P2CS. 7 Furthermore, there are numerous sensors with periplasmic, membrane-embedded, and cytoplasmic sensor domains and a great diversity of regulator protein contexts.
TCS Rewiring by Changing Residues in Sequences
Sequence mutations change sensors and regulators, for instance the specificity of the stimulus recognized or the genes regulated. To gain concrete information useful for engineering, we looked closely at sequences from several bacterial model organisms, focusing especially on the recognition site and the DNA and promotor binding sites. Annotated information on these signatures is often not available and hence relies on detailed manual annotation as well as sequence comparisons. We revalidated predictions by extensive sequence-structure comparisons (more information see Supplementary material).
TCS Stimulus Signatures
We annotated here several stimulus recognition sites in different model organisms (E. coli 536, E. coli CFT073, E. coli K12 W3110, E. coli O157:H7 EDL933, E. coli K12 MG1655, E coli O157:H7 Sakai pO157, E. coli UTI89, Salmonella, Bacillus subtilis, Staphylococcus aureus, Legionella pneumophila, Listeria monocytogenes, Pseudomonas aeruginosa, and Mycoplasma pneumoniae) and for different stimuli (Table 1A; phosphor, iron, copper, osmotic, stress, citrate, fumarate and nitrate/nitrite;23–25 sequence, genome and domain analysis, see Materials and methods). Table 1A shows the best consensus derived. However, for concrete engineering experiments and detection in new genomes, the signatures themselves are important and are given in detail summarizing all investigated sequences. They can be used directly for engineering. Detailed alignments are given in Supplementary material, section 1.2.
Stimulus recognition consensus sequences for various TCS stimuli.

Only the consensus recognition sequences are listed according to Uniprot. Well annotated sensors and organisms were compared as listed in Supplementary material. The sensor protein recognition site composition depends on the signal and is independent of the organism. Exact sequences and positions are aligned in Supplementary material. Accurate numbering according to E. coli proteins can be transferred to other organisms. Conserved amino-acids are labeled in bold print. Less conserved amino-acids are labeled in lowercase.
For rewiring, the transfer of such consensus sequences should be possible between organisms and proteins with the same sensor. To test in how far this is possible, we compared in detail the nitrate/nitrite recognition site (nitrate/nitrite sensor proteins NarX and NarQ; Table 1B). For different sensor proteins in the above-analyzed organisms, the structure of the sensor is accurately known (NarX or NarQ). We compared these sensor sequences in several E. coli, Salmonella, Vibrio and Haemophilus influenzae strains. The critical sensory region identified by sequence analysis was comparable in spite of the two different organisms and different proteins (for NARQ_ECOLI periplasmic region: position 35–146; numbering according to the E. coli Uniprot sequences). This supports the hypothesis that the signal is much more important than the organism or even the TCS family. In general, the recognition sites seem to depend strongly on the signal type, but remain conserved across the tested species.
Alignment of the Nitrate/Nitrite recognition site comparing NarX and NarQ. 1

For the same signal, two different sensors are compared in several E. coli, Vibrio, Haemophilus influenzae and Salmonella species regarding the Nitrate/Nitrite binding site: We identified the critical region for sensoring by structure analysis of the periplasmic region (NARQ_ECOLI periplasmic region, position 35–146). Subsequently different protein sequences and organisms were compared. The completely conserved sequence parts (indicated by stars) support that the sensor sequence depends more on the signal and not on the protein or organism type. Colon and point indicate well and less well conserved amino acid positions.
Binding Sites on the DNA
Another way to modify TCS functionality is to exchange the cellular response. Therefore, we analyzed the DNA binding site between regulator protein and DNA. Promotor information is normally badly annotated. The required promotor data retrieval in this study was achieved in a manual, hand curated manner by direct sequence comparison. DNA binding sites for target genes in E. coli K-12 were first collected from different sources (Prodoric, 26 DBTBS, 27 TractorDB, 28 and PDBSum) and afterwards analyzed applying specific perl-scripts and regarding further E. coli strains (E. coli 536, E. coli CFT073, E. coli K-12 W3110, E. coli O157:H7 EDL933, E. coli K-12 MG1655, E. coli O157:H7 Sakai pO157, E. coli UTI89). Conserved motifs for the DNA binding sites were summarized in form of consensus sequences per TCS family (E. coli, Table 2A; other gram-negative bacteria, Table 2B). Re-annotation using databases and subsequent sequence analysis tools are described in Materials and methods.
Specific target gene DNA sequences in E. coli. 1

Profiles of target gene binding sites bound by regulators in E. coli are given. Consensus sequences were derived from detailed multiple alignments (see Supplementary material) mining several databases (Prodoric, TractorDB, PDB and PDBSum, PubMed). Sequences and positions were aligned (Supplementary material). Given binding sequences were first found in E. coli K-12 strains and were verified for the other E. coli strains (see Supplementary material) using motif specific scripts (Materials and methods). Less conserved parts are labeled in lowercase letters, motifs with brackets and strongly conserved parts are highlighted by black boxes.
Specific target gene DNA sequences in further gram negative bacteria. 1

The table shows TCS target gene promotor sites in Salmonella (two strains) and Shigella. Capital letters indicate similarities within the binding site between the three compared organisms.
In most cases the promotor nucleotide sequences identified were quite short. As analyzed previously for different promoter sequences,29,30 we found that the TCS promoter sequences we identified have to occur in multiple copies to allow for higher specificity (including different affinities and different functions). Motifs were often repeated allowing oligomeric binding of the regulator protein.
Based on our analyses, it was possible to retrieve the concrete numbers of replicates and distances between the replicates: Table 3 summarizes the regulator proteins, the regulated genes, the numbers of binding site replicates, and the distances between the replicates.
Promotor binding sites.

As these results show that the stimulus recognition sites and promoter regions are well conserved, we are confident that the resulting consensus sequences given in Tables 1–3 will be of great help in direct design experiments 17 (see also Supplementary material, Figure S2 and Table S2 for detailed suggestions on HisKA substitution design).
TCS Rewiring by Domain Shuffling and Diverged Domains
The screens furthermore revealed more extensive changes in TCS, such as domain swapping. We identified diverged regulators or sensors in a genome where only one partner is known (Legionella, Listeria) and spot strongly diverged TCS by conserved domains in a new context (several examples including M. pneumoniae).
Diverged TCS domains
Extensive sequence analysis per TCS family, including related organisms, enabled us to better describe and predict the regulatory function for three TCSs in L. pneumophilia. New partners could be found for the osmosis-sensing family (OmpR) and the nitrate/nitrite response family (NarL). Table 4A contains the predicted and previously missing partners, the identification methods, and the TCS functions. Regarding the organism L. monocytogenes, three new TCSs within the NarL and the OmpR family could be identified, see Table 4B.
Recognition of divergent TCS and missing TCS partners.

New annotated features (interactions or part of TCS) apparent from sequence searches with various available TCS sequences and domains in the genome sequence (Genbank acc. No.: AE017354, Chien M, et al, 2004). Regulated proteins are given as well as homologous standard TCS. Predicted changes (mainly by their operon context) in their function for L. pneumophila are indicated on the right. The right-most column summarizes which aspect of the TCS is reported here new.
Listed are well characterized homologs from other organisms which have the same function within the same family.
Table contains additional features (interactions or parts of TCS) extending what is already known in KEGG or annotated in Genbank (Acc. No.: AE017262) or Listilist (http://genolist.pasteur.fr/ListiList/). On the left the TCS family is given. Starting from B. subtilis TCS sequences we searched for missing sensor and regulator proteins. The right-most column summarizes which aspect of the TCS is reported here new.
Some of the identified proteins are already known to be involved in TCS, but their connection to a specific family is unknown. The now identified TCS partners are critical for the functioning of these TCS in Legionella and Listeria. They justify further analysis and confirmation by direct experiments.
Extensive TCS Domain Shuffling
Further divergence may lead to the appearance of typical TCS domains in a new context. To detect such domain shuffling events, we applied PROSITE predictions, further sequence analyses, and literature mining. All examples investigated scrutinized proteins with either a HisKA domain or a RR domain, focusing on rather diverged cases. Four prokaryotic and even three eukaryotic examples are shown with far diverged proteins including new functional properties (Table 5). Two biotechnologically interesting examples are described in more detail:
Natural examples for domain shuffling in divergent TCS. 1

The table shows natural domain shuffling events where sensor domains and response regulator domains appear in different new contexts. In the three prokaryotic as well as in the eukaryotic examples only domains can be recognized but new functions are adopted.
Shuffled sensor domain: The branched-chain alpha-ketoacid dehydrogenase complex (BCKD) in mice was considered as a quite diverged example. 31 BCKD possesses a characteristic nucleotide-binding domain and a four-helix bundle domain similar to a TCS sensor. Binding of ATP induced disorder to ordered transitions in a loop region at the nucleotide-binding site. These structural changes led to the formation of a quadruple aromatic stack in the interface between the nucleotide-binding domain and the four-helix bundle domain, finally resulting in a movement of the top portion of two helices and to a modified enzyme activity. Our analysis indicates a diverged TCS with HisKA domain but without an RR domain and with new cellular response, namely to change enzymatic activities. Until now only the structural similarity to the Bergerat fold family has been demonstrated by inhibition experiments using radicicol as an autophosphorylation inhibitor for histidine kinases 32 but there is no in vivo evidence of BCKDHK in a signaling event of a two-component histidine kinase. In contrast, two component systems in plants such as maize seem to be genome-wide spread 33 (see Supplementary material, Table S3).
Shuffled regulator domain: If further signaling is mediated by transcription, the trans-activation domain involves a wide-range of different DNA binding motifs. Such domains appear also in new enzyme contexts or activities. One identified eukaryotic example for natural domain shuffling of a RR domain in a new protein context was the predicted serine/threonine protein kinase ppk18 in the “fission yeast” Schizosaccharomyces pombe. Ppk18 plays pivotal roles in cell proliferation and cell growth in response to nutrient status. 34 A RR domain is located C-terminal in the protein (well conserved PROSITE signature PS50110) and is target of rapamycin (TOR). TOR itself activates ppk18 by phosphorylation but does not contain the typical HisKA domain. Consequently eukaryotes can have similar operational interactions as typical prokaryotic TCS, in particular in yeast and in plants. Our computational analysis of this protein function according to the available data suggests a rather similar operation according to its interactions, in particular by its involvement of a RR domain (see Supplementary material Table S4).
High divergence is easily achieved by new molecular partners of the domain that is known from prokaryotic TCS, as shown in these eukaryotic examples. Nevertheless, there is a certain level of convergent evolution observable in the examples, regarding their regulatory function and effect.
A Putative new Family of TCS in Mycoplasma Pneumoniae
Modification in TCS can even go so far that both TCS partners are quite diverged and it is difficult to identify them as TCS. Combining bioinformatical sequence and structure analyses, there is a chance to identify such (quite) degenerated TCS in prokaryotes. A putative new TCS family encoded in the M. pneumoniae genome, so far described as TCS-free, is suggested here. In particular, MPN013 and MPN014 could form a rather diverged sensor and regulator pair in M. pneumoniae.
Putative Sensor: These proteins could not be identified with simple sequence searches, since direct sequence similarity searches did not yield significant hits. 35 After at least seven PSI-BLAST iterations, the collected alignment included described TCS sensors in addition to the UPF family to which MPN013 was previously known to belong to, the non-annotated protein family DUF16 exclusively found in Mycoplasma.
To verify MPN013 as a potential sensor protein structure, analysis with respect to the primary, secondary and tertiary structure and several alignments were established: A re-check of the prediction via PSI-BLAST analysis identified M. pneumoniae protein MPN013 as a potential sensor protein; its primary structure sequence was similar to NarX in Psychrobacter arcticum (PSI-BLAST e-value 6 × 10–13 after 5 iterations).
Afterwards we analyzed the secondary and tertiary structure of MPN013. The homology model applying SWISS-MODEL yielded the template 2ba2A (crystal structure of MPN010, another member of the DUF16 family) for MPN013. 2ba2A is a four alpha helix-bundle corresponding to the HisKA domain of a sensor protein. The MPN013 sequence extended the C-terminus and contained an additional second domain.
MPN013 starts as all sensor proteins with an unspecified domain (1–120) probably representing a signal-perception domain. Following this, we found an alpha-helical structure (130–165). This outcome was supported by secondary structure prediction (PredictProtein 36 and Predator 37 ) and was in line with the homology model. The last part was a mixture composed of helices, sheets, and loops. Secondary structure predictions were not completely identical. However, secondary structure alignments with the software SSEA 38 showed a similarity to alpha/beta sandwiches (z-score 2.28; normalized score of 54.5).
To further verify the features required for a TCS, it is demonstrated that MPN013 can be aligned in primary and secondary structure with NarX from Psychrobacter arcticus (Fig. 1). The corresponding E. coli NarX sensor was added for comparison purposes. The structure (Fig. 1; top panel) was given according to the structure template 2c2a (HisKA853 of Thermotoga maritima) from PDB, which should be valid for NarX as well as HisKA in general. Conserved residues for TCS are highlighted (yellow boxes) and the homology model for MPN013 (PDB entry 2ba2_A for MPN010) is shown in green.

Divergent TCS sensor in M. pneumoniae.
Four conserved amino acid boxes were analyzed next: The first box (Fig. 1, yellow) represents the strongly conserved histidine environment, which binds phosphor for the transfer to the RR. This site is situated in the four-helix bundle. The comparison between the E. coli, P. arcticus and MPN013 sequences already made clear that this site was variable with respect to its position and environment. The secondary structure comparison revealed that the histidine has to be situated at the end of an alpha helix. However, the further environment of the histidine residue in MPN013 is diverged. A second box could mainly be found in E. coli and was therefore rarely conserved. The third and fourth conserved boxes comprise the ATP-binding site (Fig. 1). Those two sites are more highly conserved, as demonstrated by the conserved PFAM based pattern Glu/Asn-X-Ile/Leu-X-Asn/Ala-X and Asp/Glu-X-Gly/Ser-X-Gly/Glu-Ile. This secondary structure comparison showed that the structure might be even more flexible than initially assumed.
Furthermore, regarding a tentative ATPase activity predicted by the sequence analysis, close comparisons with the HisKA subclasses as described by Grebe 3 showed that the MPN013 histidine environment was new (see Supplementary material). It was clearly different than what has been already described; however, the closest relative was a mixture of the HK3b and HK11 environment. An autophosphorylation region was identified and contained the conserved amino acids histidine and arginine just as in the HPK11 family. Within the ATP binding site, the MPN13 motif contained the conserved glycine as observed in the HK3b motif.
Consequently, even when the overall structure of the putative sensor did not match perfectly, conservation was apparent in structure as well as with respect to key residues. However, other parts of the sequence vary more than standard TCS, which explains why this was not detected by sequence comparison before. Furthermore, though key conserved structure and sequence features point to a diverged TCS in M. pneumoniae, its divergence may lead also to diverged function (see examples above).
Putative Response Regulator: Additional predictive evidence for this diverged TCS became available by searching for a corresponding regulator protein:
This search was initiated by an organism specific iterative BLAST with NarL from P. arcticus. NarL is the corresponding RR to the HisKA of NarX in P. arcticus, which was the most similar HisKA to MPN013. Consequently, on a primary structure level, NarL is similar to the Mycoplasma protein MPN014. This result was further supported by gene neighborhood considerations,39,40 which are also expected for TCS as sensors and regulator genes are often situated directly next to each other in different genomes. 41
In order to test this hypothesis on a secondary structure level, a homology model for MPN014 was calculated. MPN014 was not only located next to MPN013, but the secondary structure sequence alignment showed that it was homologous to NarL from P. arcticus and the general structure template 1p2f (TM_0126 of T. maritima) for RR in TCS. It has already been noted that MPN014 contains a topoisomerase/primase domain (“toprim” domain) including a nucleotidyl transferase or hydrolase function according to PFAM. 42
For a detailed structure sequence comparison the secondary structure is provided (according to the PDB file: 1rnl) and the sequence of NarL in E. coli. A comparison between the MPN014 sequence and NarL in P. arcticus is shown in Figure 2. The sequence comparison displayed good similarity between NarL in P. arcticus, NarL in E. coli and MPN014 in M. pneumoniae (conserved residues are highlighted).

Diverged TCS regulator in M. pneumoniae.
The phosphor binding alpha/beta 3-layer sandwich was apparent (red letters in the NarL sequence) as well as the DNA-binding alpha-orthogonal bundle (blue letters). The alignment was good enough to enable identification of all conserved regions (colored boxes). The second part of MPN014 did not display an HTH motif, but the similarity of MPN014 to the topoisomerase/primase domain and its particular relatedness to DNA-primase related proteins (protein cluster CLSK542094) supported the idea that the topoisomerase/primase domain may bind to DNA (just) as many regulators in TCS do.
Based on the patterns, which were only partially conserved, it became apparent that this element was probably a quite diverged RR. (i) The sequence contained only weak hydrophobic residues in the region corresponding to beta-strand-1. (ii) Immediately following, it contained the conserved pair of acidic residues involved in binding the metal ion for phosphorylation reactions, it was the combination glutamic acid plus glutamine as second amino acid. (iii) Hydrophobic residues corresponding to beta-strand-3 and the immediately following absolutely conserved aspartic acid that is the site of phosphorylation were observed, as well as some hydrophobic residues corresponding to beta-strand-4, but the sequence did not contain the immediately following and highly conserved serine/threonine that binds to the phosphoryl group and mediates conformational change. This was replaced by an asparagine.
Nevertheless, based on the above results, we see that structure and sequence features are sufficiently conserved to suggest that the pair MPN013/MPN014 could be a rather diverged TCS. Furthermore, its diverged functionality is at least used by M. pneumoniae (expression data see below).
The entire DUF16 family is M. pneumoniae specific, but contains a number of potential sensor proteins (MPN139, MPN138, MPN137, MPN130, MPN127, MPN104, MPN038, MPN013, MPN010, MPN655, MPN524, MPN504, MPN501, MPN410, MPN368, MPN344, MPN287, MPN283, MPN204), and the encoded two M. pneumoniae proteins related to the DNA-primase family could act as potential regulator proteins (MPN014, MPN353). In M. genitalium we have only identified a homologous counterpart for the regulator. However, the multiple copies found are another indicator that the protein family is at least useful and kept in M. pneumoniae (and this although in general there is genome reduction in parasite genomes). This is further confirmed by EST expression data for MPN013 and preliminary expression data for MPN014 (see http://coot.embl.de/Annot/MP/).
Rather diverged TCSs do thus occur in various and quite different instances. They are involved in changing of partners, but also in changing of different residues, cooperative changes can even lead to the adoption of new functions. This is difficult to design. For such experiments, complex, correlated changes in the overall protein structure and function revealed eg, by statistical coupling analysis 43 have to be taken into account. This method has been shown to work well for the redesign of proteins such as Hsp70 and of allosteric changes. 44 A key requirement is a sufficient statistical sampling, ie, large alignments to study sequence variation in the protein family of interest. Furthermore, extensive structural information is required. 45 Combining both aspects allows defining specific and important regions within the protein where mutations influence each other. However, for large protein families these regions predict quite well coordinated or cooperative changes in proteins. 43 This can then be exploited for protein design, for instance the design of protein chimeras while preserving functionality of critical domains. 46 We are confident that this approach will also work for two-component system design and maybe even in a diverged TCS. At least a sufficient number of TCS sequences, required to get the statistical power for reliable predictions, are available as well as known structures to define structural sectors of conserved and cooperatively changing regions in two-component systems for sensor and regulator proteins.
TCS Rewiring by Additional Components
TCS can furthermore be modified by additional components, so-called connectors. These modify or enhance signal transmission, increase the binding to regulator proteins, or act as additional response modifying proteins within a TCS.47,48 Such interacting proteins enhance evolution and adaptation of TCS further and are also an interesting option to modify their rewiring. In general, the connector is present in addition to the sensor and regulator protein.
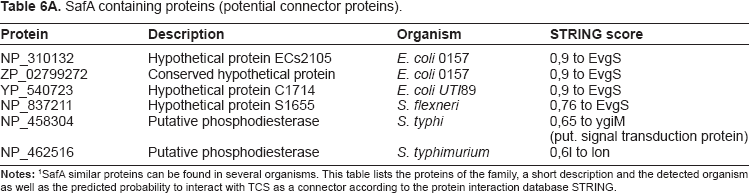
Connector family SafA, Sensor-associating factor A: Eguchi et al describe the SafA as a small membrane protein in connection with TCS, to be found in the EvgS/EvgA and PhoQ/PhoP TCS in E. coli. 48 The expression of EmrY is induced by activated EvgA. The activated EvgS/EvgA system activates the PhoQ sensor protein of the PhoQ/PhoP. SafA thus supports the interaction between the two TCS.
With the help of organism specific alignments, sequence and gene context analysis, it could be confirmed that SafA does not only occur in E. coli but also in Shigella and Salmonella. All identified potential SafA proteins are unknown or hypothetical proteins and STRING predicts interactions to either EvgS or proteins with similar functions (see Table 6A and Supplementary material, Table S5).
SafA containing proteins (potential connector proteins).
EAL and GGDEF domains: EAL domains have diguanylate phosphodiesterase activity and are found in diverse bacterial signaling proteins.49,50 If they interact with a TCS, they may influence it. This is documented for GGDEF domain containing regulators in many prokaryotic signal connected proteins, as the GGDEF domain has an enzymatic activity for synthesis of the second messenger molecule cyclic-di-GMP. 51 We looked for new examples applying gene context methods, literature mining, and the STRING database. 39 Table 6B displays the predicted interaction partners for several proteins containing an EAL-domain. Indeed, EAL proteins were often predicted to interact with known regulator proteins or had partners with DNA-binding domains (as most of the known RR in TCS). Alternatively they interacted with proteins containing the GGDEF domain. EAL and GGDEF domains can frequently be found in response regulator domain containing proteins.
Putative connector proteins containing an EAL-domain and their interaction partners.

Interaction predictions included sequence- and structure analysis and data from public interaction databases such as STRING database.
For protein engineering or synthetic biology experiments, connectors could be used to specifically modify TCS or connect two TCS. The analyzed examples are known and shown to work in several organisms, but the connector may also be tried on TCS from other species by just over-expressing these together. Evolution uses a large pool of potential interacting proteins.52,53 The same connectors are used only on comparatively short distances: In prokaryotes in particular, there is a counter selection, as wrong interactions lead to wrong regulation. However, as in eukaryotic evolution, where new protein interactions compensate for random drift in functional complexes, 54 new protein design may of course adapt connectors for broader use. For instance, the SafA connector protein family efficiently bridges two different TCS systems. This can be attractive for new designs in synthetic biology such as synthetic circuits. 55
TCS can also occur in eukaryotes such as plants, for instance in maize 56 and in Arabidopsis, where systems showing activities similar to TCS are found.57,81 These could in principle be quite diverged eukaryotic TCS, similar to the Mycoplasma example, or fairly close to standard TCS. Supplementary material, Table S6 shows both is true to some extent. Thus, in maize 25 proteins similar to HisKA proteins could be found, but only 20 of them are known to be involved in a plant TCS; for Arabidopsis the ratio is such that from 61 proteins similar to HisKA proteins there are only 16 proteins known and annotated to be participating in a TCS. For response regulators the differences between identified domains and annotated response regulators are even larger, indicating more divergence. However, this analysis also shows that a considerable number of these TCS are surprisingly well conserved in their domain architecture, and sometimes even in their motifs and signatures. At least these comparatively conserved eukaryotic TCS can be tackled with the strategies and bioinformatics data given here based largely on prokaryotic data. For more diverged eukaryotic TCS again careful and complex calculations as outlined above are the only potential strategy. However, the number of eukaryotic TCS sequences available is comparatively low and hence the statistical power of sequence-structure correlation algorithms will not be strong.
The various examples and three modification strategies applied also raise the question about a quantitative estimate of TCS divergence in general. To answer this question we first give an overview and a sequence tree on the species distribution of HisKa and response regulator domains in general (see Supplementary material, Figure S1). Furthermore, we made a detailed quantitative assessment of TCS divergence regarding the HisKA site (see Supplementary material, Figure S2) and performed various analyses about the different context in which TCS domains can occur. Those analyses included the frequency of different domain-family occurrences as well as specific domain combinations (Supplementary material Table S1 gives a detailed example). However, to get a more general overview, we give in Table S6 also an estimate on the occurrence of key TCS domains versus the number of annotated and known TCS in several bacterial genomes plus the recent data on maize as well as Arabidopsis plant genomes. As the data show, the number of domains is in all cases clearly higher than the number of annotated TCS. These new domain contexts for key marker domains of TCS give an upper bound on the number of highly diverged TCS for these different species, in reality the actual figure is lower (depending on how strict the function of the TCS as a sensor plus phosphorelay system is defined).
Conclusions
The plasticity of TCS is of high interest. It has been studied since a long time and documented in various databases.4–6 The aim of this study is to identify evolutionary modification scenarios and analyze their use for engineering TCS. Extensive genome comparisons, sequence, and structure analysis of natural instances revealed three general rewiring scenarios modifying TCS: (i) exchanges of few amino acid residues or (ii) of whole domains, 54 as well as (iii) applying connector proteins.47,48,50 For engineering, the accurate and specific binding sites, promoter motifs, and stimulus recognition motifs described should work best. In contrast, the identified diverged TCS, including potential eukaryotic variations, partners for Listeria and Legionella TCS, and a highly diverged TCS family in Mycoplasma show that extensive changes in TCS function are possible, but involve complex cooperative changes, which are not easily predicted or designed. Of the connectors analyzed, the SafA family may be attractive for synthetic circuit design, 55 as they efficiently bridge TCS systems.
Materials and Methods
The identification and analysis of individual TCS components was performed in separate steps and with specific methods for sequence alignment, for the investigation of domain and structural features, for their gene context, as well as for pathway aspects.
Methods for Sequence Analysis
Large-scale screens for diverged TCS were conducted on different databases (PFAM, 21 the protein database Uniprot 22 ) and we examined further repositories such as MIST2, 4 SENTRA 6 and P2CS. 7 Furthermore, KEGG 58 databases as well as specific sequence searches were used to collect all known and available TCS in standard model organisms. Iterative sequence searches and domain analyses were conducted as described previously. 40 We included the following model organism and strains: E. coli genome sequences E. coli 536, 59 E. coli CFT073, 60 E. coli K-12 W3110, 61 E. coli O157:H7 EDL933, 62 E. coli K-12 MG1655, 63 E. coli O157:H7 Sakai, 64 E. coli UTI89 65 as well as Shigella 2a str. 2457T and Salmonella typhi strains CT1866/Ty267 ATCC 700931; S. typhimurium LT2, 68 B. subtilis (strain 168), S. aureus (COL), 69 L. pneumophila (Philadelphia 1),70 L. monocytogenes (EGD-e71/F236572) and M. pneumoniae (M129) 73 as well as all sequences and organisms available from PFAM. Data on promotor interactions were retrieved from the ProDoric database, 26 which comprises information from exhaustive literature analyses, computational sequence predictions, and DBTBS, 27 a reference database of published transcriptional regulation events on B. subtilis. This source of information was complemented by studies performed in TractorDB, 28 which contains a collection of computationally predicted transcription factor binding sites in gamma-proteobacterial genomes.
Domains were tested and verified by comparison with known domain families, including data from databases such as SMART, 74 PFAM, 21 and Uniprot. 22 TCS components of various genomes were extensively compared in their sequence composition, intrinsic properties, as well as regarding amino acid conservation and variation.
To calculate consensus sequences, the
The DNA binding sites in related genomes were identified with perl-scripts, which employ the Fuzznuc program of the EMBOSS package 77 as a method for pattern searching. A binding site was assigned as soon as it matched the pattern. Screening runs allowing mismatches were also conducted and results were manually annotated, eg, whether the pattern was long enough to tolerate mismatches or whether symmetry-breaking mismatches were not tolerated. The described approach enabled the identification of conserved binding sites with mismatches in related E. coli genomes starting from E. coli strain K-12.
Methods for structural analysis
Based on results from PFAM and SMART, a search for essential functional domains in TCS was initiated. Moreover, an analysis of their cellular location within the cell using annotation from literature and public databases was performed.
To determine domain boundaries, we included functional and structural information. The transfer of domain features to non-annotated proteins was achieved with the help of search patterns (according to PROSITE and PFAM patterns).
After domain analyses individual domain results were assembled to a complete protein structure. Tertiary and secondary structure information was added from PDBSum, AnDOM, SCOP 78 and CATH. 79 Homology models were created using SWISS-MODEL. 80 Further analyses included secondary structure, binding features as well as function-specific motifs and key conserved structural residues. The structure of TCS was furthermore analyzed in more detail starting from available PDB structures. 81 We started with well-annotated domains in sensor and regulator proteins and compared these to less well-characterized sequences. Furthermore, detected structural or sequential characteristics in all analyzed proteins were transferred to proteins without annotations.
Structure predictions were performed by PredictProtein, 36 and Predator. 37 Secondary structure alignments were derived with the Server for Protein Secondary Structure Alignment (SSEA). 38 Predictions for protein interactions exploited the STRING tool, 39 structure analyses, and literature mining.
Author Contributions
BK implemented the process concept and alignments. BK, TF, and JB programmed perl-scripts and calculated all data. JB, RG, FF, TD and BK analyzed data and participated in writing the MS. TD led and guided the study and supervised BK, TF and FF. All authors approved the final version of the MS.
Funding
We thank German Research Foundation (TR 34/A8 in particular as well as TR 34/Z1; Da 208/13-2, SFB 479) for support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors declare there are no competing interests.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgments
We thank German Research Foundation (SFB 479, Da 208/13-1, TR 34/A5/Z1) for support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We thank Dr. Ulrike Rapp-Galmiche for stylistic and language corrections.
Supplementary Data
Supplementary material contains sequence data and alignments as well as the analysed HisKA families.