
Abstract
The exact mechanisms of prion misfolding and factors that predispose an individual to prion diseases are largely unknown. Our approach to identifying candidate factors in-silico relies on contrasting the C-terminal domain of PrPC sequences from two groups of vertebrate species: those that have been found to suffer from prion diseases, and those that have not. We propose that any significant differences between the two groups are candidate factors that may predispose individuals to develop prion disease, which should be further analyzed by wet-lab investigations. Using an array of computational methods we identified possible point mutations that could predispose PrPC to misfold into PrPSc. Our results include confirmatory findings such as the V210I mutation, and new findings including P137M, G142D, G142N, D144P, K185T, V189I, H187Y and T191P mutations, which could impact structural stability. We also propose new hypotheses that give insights into the stability of helix-2 and -3. These include destabilizing effects of Histidine and T188-T193 segment in helix-2 in the disease-prone prions, and a stabilizing effect of Leucine on helix-3 in the disease-resistant prions.
Keywords
Introduction
Misfolding of the prion protein (PrP) is believed to be responsible for the Transmissible Spongiform Encephalopathy (TSE) diseases (Prusiner, 1998). Experimental investigations suggest that the pathogenesis of TSE is characterized by the unfolding of the normal Prion protein (PrPC) followed by misfolding into an infectious “scrapie” isoform (PrPSc) (Pan et al. 1993). According to the protein-only hypothesis, PrPSc promotes structural conversion of the cellular PrPC into the pathogenic conformation (Prusiner, 1998; Prusiner et al. 1998). The pathogenesis presumably involves the initial formation of PrPSc, which is a result of a point mutation(s) or some exogenous factors, and which subsequently interacts with and converts PrPC molecules into PrPSc molecules. The last decade of research has provided a significant amount of evidence that supports this hypothesis (Mead, 2006).
Known PrPC structures reveal that the C-terminal domain (positions 125 to 230) is structured and contains three α-helices and a short β-sheet that includes two strands (see Fig. 1), whereas the N-terminal domain (positions 23 to 126) is highly flexible and cannot be assigned a particular conformation (Riek et al. 1997; Riek et al. 1998; Lopez-Garcia et al. 2000). At the same time, the structure of the PrPSc isoform is currently still unknown.

Sequence and mutations in the C-terminal domain of huPrP together with the ribbon drawing of the corresponding 3D structure (positions 125 to 228 of 1HJM). The secondary structure segments are denoted by underscores.
Spectroscopic studies have shown that PrPC is composed of about 42% α-helices and 3% β-sheets, whereas PrPSc is composed of only 30% α-helices and 43% β-sheets (Pan et al. 1993). Thus, the conformational transition of PrPC into PrPSc has to involve unfolding of some -helices and formation of new β-sheets. Helix-1 is the most conserved in PrP sequences and forms only a few interactions with the rest of the C-terminal domain. These facts led to a model in which helix-1 was considered as a starting point for conformational transition and forms a β-like aggregate, whereas helix-2 and helix-3 retain their conformation (Huang et al. 1995; Morrisey et al. 1999; Wille et al. 2002). Some recent models of the pathologically misfolded form of PrP also show that the helix-1 region is unstable and has to unfold during the conformational transition (Eghiaian et al. 2004). At the same time, recent results provide strong evidence that helix-1 is not converted into a β-sheet during the aggregation of PrPC to PrPSc (Watzlawik et al. 2006). This conclusion is also supported by experimental data obtained using low-resolution electron crystallography which suggest that helix-1 in PrPSc refolds into a left-handed β-helix (Wille et al. 2002), while subsequent work shows that helix-1 is not included in the β-helix but forms an unstructured loop (Govaerts et al. 2004). These discrepancies motivate this work, in which we use sequence based analysis to find factors that could impact the stability of particular secondary structure segments.
A number of point mutations in the human prion have been identified. A significant proportion of all mutations are found within the structured C-terminal domain; 27 out of total of 30 as reported in (Kovacs et al. 2002) and 37 out of 55 as reported in PrionDB at http://www.receptors.org/Prion/ (Horn et al. 2001). Thus, we focus our attention on the C-terminal domain (see Fig. 1). Pathogenic mutations are classified based on their association with prion diseases that include Gerstmann-Straussler-Scheinker disease (GSS), Creutzfeld-Jakob disease (CJD), and Fatal Familial Insomnia (FFI). The number of possible single-point mutations in the C-terminal domain is relatively large (109 positions * 19 = 2071), and thus it is not feasible to physically check every one of them using wet-lab techniques. Well-designed computational experiments (such as the design we propose) can reveal promising candidate factors, which serve as new hypotheses for wet-lab investigation. To this end, another of our goals is to use sequence based analysis to find point mutations that could predispose PrPC to misfold into PrPSc.
In contrast to other sequence analysis based approaches that contrast prion proteins with structurally similar proteins such as Doppel (Kuznetsov and Rackovsky, 2004), we present a novel in-silico approach based on the assumption that some species are susceptible and others are resistant to prion disease (PD). We divide the available prion sequences from vertebrate animals into those that are prone to PD, and those that are apparently resistant, i.e. there are no reports of any known PD in that species and research suggests that they do not develop PD. We then compare the PrP sequences from these two groups (hereafter “the contrasts”), with a focus on the C-terminal domain. To the best of the authors’ knowledge, only two prior sequence-analysis-based contributions perform similar contrasting analysis, but they focused on identification of β-aggregating stretches (Tartagia et al. 2005) or contrasted just four prion proteins (Pappalardo et al. 2007). We used an array of computational techniques including multiple sequence alignment, exchange group similarities, and feature selection methods to identify possible factors that distinguish the contrasts for a larger set of 11 proteins. We suggest that such discriminating factors are potentially important in the conformational change from PrPC to PrPSC. The results of this analysis are best viewed as either evidence confirming known factors associated with prion misfolding, or newly hypothesized factors that predispose the misfolding.
Materials and Methods
Dataset
We extracted the sequences of all prions that were deposited in Protein Data Bank (PDB) (Berman et al. 2000) as of September 2007. This database is expert-curated, which assures high quality of the data, and includes structural information, which allows us to identify secondary structure regions and perform structural analysis. The 70 prion sequences stored in PDB belong to 15 species: chicken (1 sequence), ovine (4 sequences), human (29), elk (1), rabbit (1), canine (1), frog (1), turtle (1), bovine (5), mouse (4), cat (1), pig (1), syrian hamster (2), sheep (5), and yeast (13). Yeast prions were removed since they have no homology with the remaining vertebrate prions, and are shown to have substantially different properties (Bousset and Melki, 2002). We filtered out redundant sequences, i.e. we selected the newest deposition for each species (except for sheep prions, for which there are two depositions from 2004; we selected the slightly older 1UW3 that does not include polymorphisms), and eliminated sequences that did not cover the C-terminal domain. We note that among the C-terminal domain sequences the four bovine sequences and the two mouse sequences are identical, while the only differences between the two sheep sequences are C148R and Q168H mutations, and among ten human prions nine sequences are identical and one differs from them by two mutations M166C and E221C. The positions associated with these mutations do not show any consistent pattern vs. our contrasts (i.e. they do not serve to differentiate PD-prone from PD-resistant species), and so the duplicate sequences are redundant and could be safely removed. It is in fact necessary to remove them; data-mining techniques such as feature selection assume that there is no redundancy in a dataset (deletion of redundant data items is a standard preprocessing step in data mining), and so the presence of redundant sequences would undermine our results.
Next, for the remaining 14 species we searched for evidence in the literature that supports existence of PD, or which suggests that they are PD resistant. Eight mammalian species (human, bovine, sheep, elk, cat, mouse, syrian hamster, and ovine) are shown to develop PD (Prusiner, 1997; Prusiner, 1998; Benkel et al. 2007; Murayama et al. 2007). At the same time, prion diseases were never confirmed for the non-mammalian species turtle, chicken and frog, and several studies suggest that they do not develop prion diseases (De Simone et al. 2006; Ji et al. 2007). For the remaining 3 species, i.e. pig, canine, and rabbit, we could not find sufficient evidence to categorize them to either class (Wells et al. 2003; Vorberg et al. 2003; Lysek et al. 2005). We note that canine shares high sequence similarity with PD-prone species, i.e. between 88% for human and 98% for cat, and moderate similarity with PD-resistant species, i.e. between 30% for frog and 41% for turtle. Similarly for rabbit and pig the sequence similarity to PD-prone species ranges between 90% (for human) and 96% (for sheep and ovine), and between 86% (for hamster) and 93% (for elk), respectively, while for PD-resistant the similarity ranges between 31% (for frog) and 42% (for turtle), and between 28% (for frog) and 41% (for turtle), respectively. It is of course possible to simply add the three uncategorized species to the “PD-resistant” class, since no evidence has been produced that they do experience prion disease. However, this would, in our view, be a serious methodological error. Our analysis contrasts species that are known to develop PDs against those that clearly do not, and this distinction directly affects all of the computational techniques (discussed below) that will be employed in our work. The inclusion of pig, canine and rabbit prions would undermine the contrasts, because we could not positively assert that these are truly PD-resistant species. Our methods are fundamentally intended to identify only those differences that perfectly distinguish between the two classes; if the classes themselves become uncertain, our entire methodology becomes merely a “shotgun correlation.” The eleven species we have selected already represent the maximal set of species that we can confidently differentiate into our two classes at the present time. It would be highly desirable to include more species in each class; data-mining techniques such as feature selection are generally intended to operate over thousands or tens of thousands of examples. Obtaining a firm determination of susceptibility to prion disease in canines, rabbits and pigs would be an excellent start.
Point Mutations
We performed multiple sequence alignment of the 11 PrP C-terminal domain sequences using ClustalW version 1.83 (Chenna et al. 2003). ClustalW produces biologically meaningful alignments that allow finding identities, similarities and differences between a set of protein sequences. Next, we searched for significant mutations based on positions that are conserved within PD-prone and PD-resistant species. Each position was categorized as follows:
Each position that includes a conserved (the same) amino acid (AA) in the PD-prone species and a conserved (the same) AA (different from the AA conserved for the PD-prone) in the PD-resistant species is categorized as significant. Such a position shows conservation within each group while at the same time it differentiates the contrasts.
Each position that has different AAs over different PD-prone species and/or PD-resistant species is categorized as insignificant. These positions show no significant conservation pattern.
Each position that has conserved (the same) AA over all PD-prone and resistant species is considered insignificant. Although these positions show significant conservation, these residues do not differentiate the contrasts.
Working from the hypothesis that TSE mutations are exclusive to PD-prone species, each significant position is a candidate factor that predisposes PrPC to misfold into PrPSc.
We repeated the same procedure using exchange groups, which represent conservative replacements of AAs through evolution (Dayhoff et al. 1978). They relax the constraint of conservation by defining equivalence classes of AAs, as derived from the BLOSUM AA substitution matrix (Henikoff and Henikoff, 1992), which in turn was derived based on the BLOCKS database (Henikoff and Henikoff, 1991). This reduces the alphabet of 20 AAs to the following six exchange groups:
Stability of secondary structure
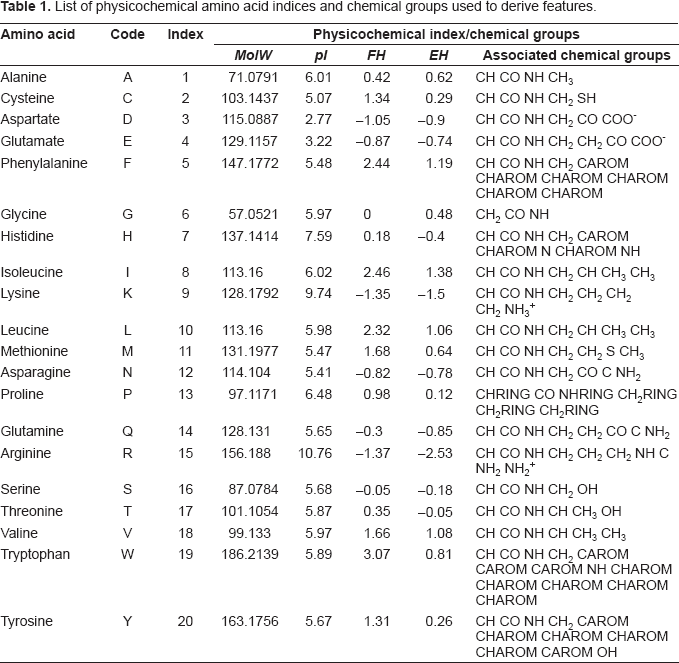
Each prion sequence was converted into a feature-based vector, and the features that differentiate the contrasts were identified using a combination of feature selection methods and correlation analysis. The features represent physicochemical properties of protein sequences that were previously used to characterize and predict certain properties related to the secondary structure of protein sequences, including structural class (Feng et al. 2005; Cao et al. 2006; Kedarisaetti et al. 2006; Kurgan and Chen, 2007) and secondary structure content (Zhang et al. 2001; Ruan et al. 2005; Homaeian et al. 2007). As such, features that discriminate between the contrasts are candidate factors that predispose β-sheet poor PrPC to misfold into β-sheet rich PrPSc. As the conformational change from PrPC to PrPSc will ultimately be driven by physiochemical properties, these features are a promising source of candidate factors. The features we analyze include:

List of physicochemical amino acid indices and chemical groups used to derive features.
Average isoelectric point, 
Composition vector, 
Order 
sum, 
where
Property groups of amino acids used to derive features.

We employed three feature selection techniques to minimize bias in our results. These are the ReliefF (Robnik-Sikonja and Kononenko, 2003), information gain (Quinlan, 1993), and the χ
2
statistics, taken between a given attribute and the binary class (PD-prone/PD-resistant). The ReliefF algorithm estimates the ability of features to separate classes. This algorithm examines nearest-neighbors of a feature vector that belong to the same or a different class as the vector under consideration. Features that categorize these nearest neighbors correctly receive a high score, and the process is repeated for each feature vector. The second selection technique is based on the concept of minimization of
Results and Discussion
Point Mutations
The aligned prion sequences are shown in Figure 2. Our analysis shows the following significant positions: 137, 144, 187, 189, 191, and 210, which are associated with the following point mutations with respect to huPrP: P137M, D144P, H187Y, V189I, T191P, V210I (see Fig. 2). Similarly, when considering conservation at the level of exchange groups, the following positions were found significant: 137, 142, 144, 185, and 187. The positions 137, 144, and 187 overlap with the results of residue conservation, while the remaining two positions are associated with G142D, G142N, and K185T point mutations. One mutation is a confirmatory result, while the remaining eight are new findings:

Results of sequence alignment between the three PD-resistant prions (top) and the eight PD-prone prions (identified by the PDB ID for the protein).
P137M (new finding). Residues that compose helix-1 are not involved in hydrogen bonds with the rest of the C-terminal domain. This is true except for Y149 and Y150 which belong to helix- 1 and whose side chain hydroxyls donate to the carboxyl groups of D202 and the CO of P137 (Riek et al. 1998). Therefore, a mutation at P137 could further weaken interaction between helix-1 and the rest of the C-terminal domain. At the same time, several studies report importance of weakened interactions between helix-1 and other segments in the C-terminal domain on the folding into a stable native structure (Hirschberger et al. 2006; Schwarzinger et al. 2006; Eghiaian et al. 2007)
G142D and G142N (new findings). A mutation at the same position, i.e. G142S, was previously classified as having a CJD-like phenotype (Gambetti et al. 2003). For this mutation, Glycine at position 142 was substituted with a polar, hydrophilic Serine. Using our approach, we identified mutations at that position involving Aspartate and Asparagine, which are very similar to each other and both also polar and hydrophilic, similar to the known mutation.
D144P (new finding). Previous research shows that D144 forms a salt bridge with H140, R148 and R208 (Zuegg and Gready, 1999). The salt bridge between D144 and R208 links helix-1 and helix-3, while the R208H mutation is associated with CJD (Riek et al. 1998). Since salt bridges are suggested to increase the stability of proteins, mutation at this position could potentially lead to destabilization of the prion's structure. Recent results also show that a point mutation leading to the disruption of a single salt bridge in p53 increases propensity to form amyloid fibrils (Galea et al. 2005).
H187Y (new finding). This position is associated with a known H187R mutation that results in GSS (Cervenakova et al. 1999). At the same time, both Tyrosine and Arginine are polar and similar in size, i.e. their van der Waals volumes are 141 and 148, respectively.
V210I (confirmatory finding). This mutation is well-known and is associated with CJD in humans (Riek et al. 1998).
We have shown that several of the new mutations we have found are closely related to known mutations involved in TSE diseases, while others may impact structural stability of the prion protein. While we were unable to find established research that would directly corroborate the remaining new mutations (K185T, V189I, and T191P), existing research indicates that mutations in this segment (which contains helix-2) may have β-sheet promoting effects. Helix-2 is characterized by a strong propensity for the extended conformation, and a single AA replacement in the vicinity of this helix is shown to significantly affect the conformational preference of the entire helix-2–helix-3 segment and to further increase the propensity for the extended conformation, facilitating conformational rearrangement in this region (Knaus et al. 2001; Kuznetsov and Rackovsky, 2004). These findings also correlate well with the high number of disease-promoting mutations in helices-2 and -3, which also points to the particular importance of these helices for conformational transition (only one disease-promoting mutation is found in helix-1 while seven and eight such mutations are found in helix-2 and helix-3, respectively).
Stability of secondary structures
Our feature selection was performed using tenfold cross-validation to assure statistical validity for our results. Features are evaluated in each fold, and then they are ranked on their performance across all ten folds. Higher-ranked features have greater discriminatory power for the contrasts than lower-ranked ones. We average the ranks reported for each feature across our three feature selection methods. We report the top five features, ordered by average rank, which have biserial correlation coefficient values >0.9 in Table 3. The biserial correlation coefficient measures correlations between ratio-scale and binary variables, and is interpreted in the usual manner (values >0.8 indicate strong correlations).
The five features in Table 3 fall into two groups: those that show higher values for PD-prone species than PD-resistant species, and those that show higher values for PD-resistant species than PD-prone species. We begin our discussion with the former group. The second feature in Table 3 is related to the composition of the N group in the AA side chains. Since N group occurs only in Histidine, this feature indicates that presence of this AA is specific to one group of prion proteins. This finding is also supported by the third feature,

Values of top five features for the 11 prion sequences: features that indicate abundance of the associated amino acids in
Top five features that differentiate between PD-prone and resistant prions.

The
In contrast, the remaining two features have higher values for the PD-resistant prions; see Figure 3B. Analysis of the aligned sequences shown in Figure 2 reveals that although Leucine is present at positions 125, 130, and 138 in both types of prions, this AA is only present in the vicinity of the C-terminal in the PD-resistant prions. As a result, positions 200, 203, and 223 (located within helix-3) were identified as significant locations based on the position-sensitive
Finally, the
Conclusions
We present a novel, in-silico approach to identify factors related to misfolding of prion proteins. We contrasted PrPC sequences of the C-terminal domains of PD-prone and PD-resistant species. The analysis focused on finding significant point mutations and investigating structural stability of secondary structures that comprise the C-terminal domain. We confirmed the V210I mutation, which is associated with CJD, and present several new findings that include P137M, G142D, G142N, D144P, K185T, V189I, H187Y and T191P mutations; destabilizing effects of Histidine and the T188-T193 segment on stability of helix-2 in the PD-prone prions; and stabilizing effects of Leucine on helix-3 in the PD-resistant species. All of these new findings are possible candidate factors that could influence conformational change from PrPC to PrPSc. They are a new set of hypotheses that should be investigated via wet-lab experimentation or (at a minimum) molecular dynamics simulations. In addition, if and when additional species can be definitively classified as PD-prone or PD-resistant, it would be quite interesting to repeat our experiments with these additional species included in the contrasts. Finally, we note that the resistance to prion diseases of the PD-resistant species could be a result of other factors besides the differences in their sequences, which should be addressed in future studies.
Footnotes
Acknowledgements
This research was partially supported by NSERC Canada, the Province of Alberta's Queen Elizabeth II graduate scholarship, and the Alberta Ingenuity Fund.