
Abstract
3-dimensional domain swapping is a mechanism where two or more protein molecules form higher order oligomers by exchanging identical or similar subunits. Recently, this phenomenon has received much attention in the context of prions and neurodegenerative diseases, due to its role in the functional regulation, formation of higher oligomers, protein misfolding, aggregation etc. While 3-dimensional domain swap mechanism can be detected from three-dimensional structures, it remains a formidable challenge to derive common sequence or structural patterns from proteins involved in swapping. We have developed a SVM-based classifier to predict domain swapping events using a set of features derived from sequence and structural data. The SVM classifier was trained on features derived from 150 proteins reported to be involved in 3D domain swapping and 150 proteins not known to be involved in swapped conformation or related to proteins involved in swapping phenomenon. The testing was performed using 63 proteins from the positive dataset and 63 proteins from the negative dataset. We obtained 76.33% accuracy from training and 73.81% accuracy from testing. Due to high diversity in the sequence, structure and functions of proteins involved in domain swapping, availability of such an algorithm to predict swapping events from sequence and structure-derived features will be an initial step towards identification of more putative proteins that may be involved in swapping or proteins involved in deposition disease. Further, the top features emerging in our feature selection method may be analysed further to understand their roles in the mechanism of domain swapping.
Introduction
Many cellular functions rely on interactions between protein pairs and are mediated by proteins in oligomeric conformations. Although there are many possible mechanisms for oligomer formation, 3D domain swapping has been proposed as an important mechanism that explains the evolution from monomeric to oligomeric proteins.1–4 3D domain swapping can be defined as a mechanism for forming oligomeric proteins from their monomers by exchanging identical or similar subunits. The swapped region can be an entire domain or a helix or β-strand or loop regions.5,6 Protein structures reported to be engaged in 3D domain swapping are distinct from the rest of the oligomers due to the signature-swapping phenomenon. Yet, they are extremely diverse based on their primary sequence and secondary structures and belong to different protein domain families and structural classes. Although domain swapping is an important mechanism for controlling multi-protein assembly, it has also been suggested as a possible mechanism for protein misfolding and aggregation.5–8 Protein structures in swapped conformations are reported to initiate pathological conformations in prion proteins and human cystatin C. They are reported to aggregate same type of proteins to generate aberrant structures.6,7,9–13 For example, amyloidogenic proteins like cystatin C and prion proteins have been shown to form dimers by exchange of sub-domains of the monomeric proteins.3,6,14 3D domain swapping phenomenon is interesting not only due to its pathological conformation factor; it is also important due to a wide range of functions mediated by the proteins in swapped conformation.7,12,13 It has been reported as a mechanism for dimer formation in odorant binding proteins6,15,16 and has also been proposed as a possible mechanism for fibril formation.7,14 Several well- studied examples for domain swapping events have been reported. For example, bovine seminal ribonuclease is a natural domain-swapped dimer that has special biological properties, such as cytotoxicity to tumor cells. 17 Barnase, a domain swapped trimer, is an enzyme that acquires enzymatic activity by cyclic domain swapping. 18 For example, Diptheria toxin, RNase, Cro (DNA repressor), Spectrin (cytoskeleton), antibody fragments, human prion protein (implicated in various types of transmissible neurodegenerative spongiform encephalopathy), human cystatin C (implicated in amyloidosis and Alzheimer's disease) and SH3 domains (important molecule in signal transduction) are shown to be having 3D domain swapped segments with crucial functional roles. 12 The functional diversity of proteins reported with 3- dimensional domain swapping is reflected in a diverse set of Gene Ontology (GO) annotations 19 obtained from PDB ID to GO annotation mapping. Table 1 is provided with the GO annotations (Molecular Function), SCOP fold and Pfam domain IDs of 10 different proteins reported with 3D domain swap mechanism along with their diverse function annotations. The study of 3D domain swapping events in proteins will be an important step towards understanding the molecular basis of the various factors that control this phenomenon and its crucial role in deposition diseases and evolution of swapping in oligomers. As 3D domain swapping is observed in different structures belong to different structural superfamilies (as an example, a set of 3 structures involved in 3D domain swapping is provided in Figure 1) with no common structural, sequence or functional patterns, identification of domain swapping events from features derived from combination of sequence and structural properties provides interesting insights into the patterns that could differentiate between the oligomers in swapped conformation and normal oligomers. In this manuscript, we report the details of a new Support Vector Machine (SVM) based classifier developed to differentiate between swapped oligomers or normal protein structures with a reliable accuracy of 73.81%. Further, the manuscript also discusses the top features emerging from the information-gain-based feature-selection method of the prediction model and its implication in large-scale analysis of 3D domain swapping in proteins.
List of 10 structures with GO annotation, SCOP fold and Pfam domain ID.


Materials and Methods
Curation of the datasets
We have performed extensive database and literature curation to collect sequence and structural data for proteins with the structural features of domain swapping. We have collected a set of PDB structures from Protein Data Bank (PDB) 20 using a combination of integrative database searches and extensive literature curation of the existence and extent of 3D domain swapping. These entries were further manually analyzed using combination of macromolecular visualization tools PyMol, 21 Rasmol 22 and literature reports. The structural entries were further processed using Domain Identification ALgorithm (DIAL) server 23 to identify probable swapped segments from the structural data. PDB ID to PubMed ID mapping and PDBSum database 24 were used to obtain primary literature reports. Since many structures are not available in quaternary state from the PDB, Protein Quaternary Structure server (PQS) 25 was consulted to obtain the quaternary assembly of the structures. From the extensive curation, 3Dswap: Knowledge-base of 3D domain swapping in Proteins, unpublished data, 315 PDB entries with 344 chains were obtained for the positive dataset. These chains were further mapped to their respective SCOP 26 folds. To curate the negative dataset, we scanned different databases (PDB, PQS, and PDBSum) for dimers or higher order oligomers that are not included in positive dataset. PDB was scanned for oligomers that are not reported to be involved in domain swapping. The negative dataset was generated after excluding the SCOP folds reported in the positive dataset. To add diversity to the negative dataset, members from a single SCOP fold was represented only once in the negative dataset. The redundant entries were removed by considering their sequence identity. Sequences extracted from structures that have >70% sequence identity were removed using the CD-HIT program. 27 We retained 213 domain swap sequences for the positive dataset. Equal number of negative data was obtained from the Protein Data Bank. The training dataset was constructed using 150 domain swapping and 150 non-domain swapping sequences. Remaining 63 domain swap sequences and 63 non-domain swapping sequences were employed for testing. Schematic representation of data curation steps, followed to generate positive and negative data, are given in Figure 2.

Schematic representation of data curation steps.
Features
The SVM model is generated using a combination of features derived from sequence, structure and physico-chemical properties. Initially, each sequence is represented by a set of 66 features. Further, a set of features that contribute to the prediction model is identified using the feature-selection approach explained in ‘Feature selection’ section. The features sets used in the prediction can be classified into three groups as sequence-derived, structure-derived and physico-chemical features.
Sequence Features
Sequence features are derived exclusively from sequence of proteins in the positive and negative datasets. The frequencies of 20 amino acids were calculated from the total number of each amino acid in a given sequence divided by protein length as explained previously in Pugalenthi and coworkers. 28 In addition, the amino acids are grouped into hydrophobic, hydrophilic and neutral amino acids (see Pugalenthi and coworkers 29 ) and the frequency was obtained for each sequence in the datasets.
Structure-derived features
Structure-derived features refer to a set of features derived from the PDB coordinates of the positive and negative datasets. Structure-based features such as solvent accessibility, secondary structures, hydrogen bonds and residue compactness were computed from the individual protein structures using JOY package. 30 Basic structure-based features used in the prediction model are overall composition of helix, overall composition of strand and overall composition of coil. Along with the generic structure-based features, we have also used ‘structure-derived fusion-features’ like hydrogen bonds in helix, hydrogen bonds in strand, and hydrogen bonds in coil where the frequency of hydrogen bonds in a given structure is coupled with secondary structure of residues that mediate the hydrogen bonds. The frequency of solvent inaccessible residues in the secondary structure classes like helix, strand and coil was also computed. Another set of structure-derived fusion-features includes the number of cysteine residues in helix, the number of cysteine residues in strand and the number of cysteine residues in coil regions. Hydrogen bonds were calculated using HBOND routine available from the JOY package. Secondary structure information was inferred using the SSTRUC program available from the JOY package. Solvent accessibility was calculated using the routine available in the PSA routine in JOY package to compute the Ooi number. Composition of secondary structural elements and frequency of hydrogen bonds mediated by residues in secondary structural elements were calculated using custom Perl scripts.
Physicochemical Features
We obtained 18 physico-chemical properties from AAINDEX 31 and its derivative UMBC AAINDEX database. 32 The computed physico-chemical properties include molecular weight, hydrophobicity, hydrophilicity, refractivity, average accessible surface area, flexibility, melting point, side chain volume, side chain hydrophobicity, polarity, heat capacity, isoelectric points and normalized frequency of α-helix, β-sheet and coil. Physico-chemical features were derived from the protein sequence of proteins from positive and negative datasets using custom Perl scripts.
Support vector machine
SVM, rigorously based on Vapnik's statistical learning theory33,34 possesses excellent generalization capability. Due to its excellent generalization capabilities, it is widely used in bioinformatics applications.28,29,35–37 When used as a binary classifier, an SVM will construct a hyperplane, which acts as the decision surface between the two classes. This is achieved by maximizing the margin of separation between the hyperplane and those points nearest in each class. Details of the formulation and solution methodology of SVM for binary classification task can be found elsewhere. 34 We provide here only final form of the decision function and the type of kernel function employed in our study.
Let xi Є RN, i = 1, 2 …, N be input feature vectors and yi Є {+1, −1} be its corresponding class label, where, N be the total number of proteins in training database. To assign a class label for a query sequence x, the trained SVM model applies the following function form:

In this equation, where, m is the number of support vectors, a subset of training dataset, m < N having non-zero positive values of the Lagrange multipliers, αi which are obtained by solving a quadratic optimization problem and b is the bias term. We have conducted our study with Radial Basis Function (RBF) kernel function defined by Equation 2.

K (xi,xj) represents Radial Basis Function (RBF) kernel. Parameter σ in Equation (2) decides the width of the Radial Basis Function kernel function.33,34 Simulations were performed using LIBSVM version 2.81 (C.C. Chang, 2001). SVM training was carried out by optimization of the value of regularization parameter and the value of RBF kernel parameter. 5 fold cross validation experiment was carried out to evaluate performance of SVM model.
Feature Selection
To identify the important features that distinguish positive and negative classes, we used Information Gain algorithm with the ranker method for the feature selection. This method was implemented using Weka 3.5. 38 The information gain for each feature was calculated and the features were ranked according to this measure.
Prediction Assessment
The prediction system is evaluated using sensitivity, specificity, accuracy, positive prediction value (PPV), negative prediction value (NPV) and Mathew's Correlation Coefficient (MCC). These measurements are expressed in terms of true positive (TP), false negative (FN), true negative (TN), and false positive (FP). The measurements are defined as follows:

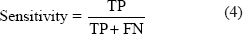




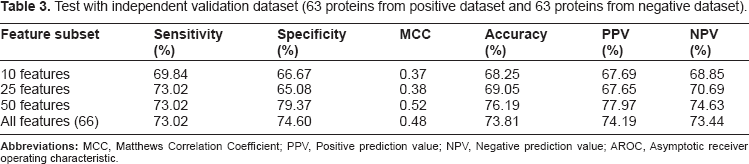
The MCC ranges from −1 ≤ MCC ≤ 1. A value of MCC = 1 indicates the best possible prediction while MCC = −1 indicates the worst possible prediction (or anti-correlation). Finally, MCC = 0 would be expected for a random prediction scheme (Matthews, 1975). Five-fold cross-validation method is also used to evaluate the performance of the model with respect to different sub-sets of the data. Results of the prediction assessment using five-fold cross validation on training dataset (Table 2) and independent validation dataset (Table 3) are provided.
Performance evaluation on training data (150 proteins from positive dataset and 150 proteins from negative dataset).

Test with independent validation dataset (63 proteins from positive dataset and 63 proteins from negative dataset).
Results and Discussion
We have developed a new SVM model to differentiate structures in swapped conformation from normal oligomers or normal structures. The model was trained on a training dataset containing 150 proteins from the positive dataset and 150 proteins from the negative dataset. The performance of the model was evaluated using the five-fold cross-validation method. As shown in Table 2, overall prediction accuracy of 76.33% was obtained by five-fold cross validation. In order to identify the prominent features, feature selection (information gain with ranker method) was performed on this dataset. We selected five feature subsets by decreasing the number of features and the performance of each feature subset was evaluated using five-fold cross-validation. As seen in Table 2, feature selection generally does not deteriorate the classification performance much until the number of features decreases to 10. Using 10 features, our model obtained 71.67% accuracy that is comparable to accuracy obtained using all features. Similar performance was observed using 25 and 50 feature subsets. This result suggests that our feature reduction approach selected useful features by eliminating the uncorrelated and noisy features. In order to examine the performance of the newly developed model, we tested our training model on the test dataset consisting of 63 proteins from the positive dataset and 63 proteins from the negative dataset. As shown in Table 3, our model achieved 73.81% accuracy with 73.02% sensitivity and 74.60% specificity using all features and 76.19% accuracy with 73.02% sensitivity and 79.37% sensitivity using 50 features. We investigated the influence of the feature reduction by plotting Receiver Operating Characteristic (ROC) curves (Fig. 3) derived from the sensitivity (true positive rate) and specificity (false positive rate) values for the classifiers using all the features and the 10 best performing features (Table 4), respectively.

ROC curves plotted utilizing the fractions of true positives and false positives values derived using top 10 features and all features.
List of top 10 selected features.

The list of top 10 features clearly indicates that features with higher classification strength are a mix of sequence, structural and physicochemical derived features. This feature distribution in both sequence and structural classes also asserts that swapping can be detected from combination of features from sequence and structural information. The 10 best performing features emerged from the feature selection using information gain algorithm offers interesting leads into the mechanism that mediate domain swapping. As no generic sequence or structure based common pattern is reported to be a hallmark of structures with domain swap mechanism, the set of top 10 features could be considered further for detailed analysis. A generic sequence or structure analysis approach could have likely missed the identification of these features, but the combination of features and machine learning based approach used in the current work enables the identification of the specific patterns between the positive and the negative datasets. Top 10 features (Table 4) identified by the feature selection method can be classified into three categories based on the mode of feature derivation. Top 10 features include four sequence-derived features (frequency of neutral amino acids, valine, tyrosine and tryptophan), one physico-chemical feature derived from sequence (refractivity), one structure-derived feature (composition of coil) and four structure- derived fusion- features (solvent inaccessible residues in coil, frequency of residues that form hydrogen bond to main chain CO in helix, number of cysteine residues in strand and number of cysteine residues in helix).
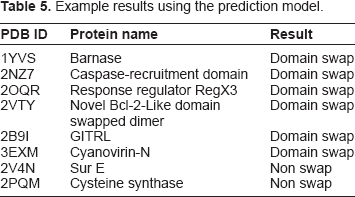
Our current prediction model has its limitations due to smaller sample size of the positive dataset. Depending upon the availability of more crystal structures with swapped conformation, the method could be improved by re-training the model using larger datasets. Due to unavailability of other methods or classifiers for the prediction of swapping events from sequence or structure data, the current method is not compared with any of the existing methods. To show the results of the prediction model, a set of example input PDB files and their respective results obtained using the current prediction model is provided in Table 5.
Example results using the prediction model.
Conclusion
Domain swapping mechanism is essential for the formation of higher protein oligomers from their monomer, protein misfolding, protein aggregation etc. Several experimental39–49 and computational studies50–56 are performed to understand various aspects of domain swapping. We have attempted to predict the phenomenon of domain swapping from the sequence and structure-derived features of a protein using machine-learning approach based on support vector machines. Identification of common sequence or structure-based features from the structures that show this phenomenon is a challenging task. We developed SVM-based classifier to predict domain swapping event using sequence and structure-derived features. This method obtained 76.33% accuracy from training and 73.81% accuracy from testing. This method could be extremely useful for the identification of domain swap phenomenon from protein structure data based on features derived from protein sequence data and structural co-ordinates. The set of features identified using our feature- selection method is providing new insights to understand a common pattern behind domain swapping and need to be explored further. The method can be improved by considering exclusive sequence based features, so that a classifier could be designed which can perform prediction using (3Dswap-pred—prediction of 3D domain swapping from protein sequence, unpublished data). Such a method could be applied at the whole genome level to scan and identify putative proteins showing domain swapping.
Footnotes
Supplementary Data
List of 66 features, positive and negative datasets (training and testing) and features derived from positive and negative dataset (training and testing) are provided as supplementary material. The supplementary data can be accessed from the following URL: http://caps.ncbs.res.in/download/3dswap_seq_struc_svm
Acknowledgements
R.S. and K.S. acknowledge National Centre for Biological Sciences (TIFR) for infrastructural and financial support. R.S. was a Senior Research Fellow of the Wellcome Trust, U.K. R.S. and G.A. thank Department of Biotechnology, Government of India for financial support. G.P. and P.N.S acknowledge the financial support offered by the A*Star (Agency for Science, Technology and Research, Singapore) under the grant # 052 101 0020. KKK acknowledges Prof. Thomas Martinetz and Dr. Stefen Moller, Institute for Neuro- and Bioinformatics, University of LÜbeck, Germany for their support.
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.