
Abstract
Understanding the structure–function relationship in proteins is a longstanding goal in molecular and computational biology. The development of structure-based parameters has helped to relate the structure with the function of a protein. Although several structural features have been reported in the literature, no single server can calculate a wide-ranging set of structure-based features from protein three-dimensional structures. In this work, we have developed a web-based tool, PDBparam, for computing more than 50 structure-based features for any given protein structure. These features are classified into four major categories: (i) interresidue interactions, which include short-, medium-, and long-range interactions, contact order, long-range order, total contact distance, contact number, and multiple contact index, (ii) secondary structure propensities such as α-helical propensity, β-sheet propensity, and propensity of amino acids to exist at various positions of α-helix and amino acid compositions in high
Keywords
Introduction
It is widely accepted that the structure of a protein dictates its function. 1 Most studies of protein structure and function rely on the analysis of the crystal structure of proteins. This is done by calculating various structure-based parameters, which have been developed to describe the folding, stability, and functions of proteins and their complexes, such as the nature of interactions among the amino acid residues and the surrounding solvent molecules, the preferred amino acid residues in the protein environment, the location of residues in the interior/surface of the protein, and the amino acid clusters. 2
These parameters focus on specific aspects of the protein structure and are described in the literature. For instance, Lee and Richards 3 developed the concept of solvent accessibility of amino acid residues. Chou and Fasman 4 studied the secondary structures of proteins and deduced the propensity of amino acid residues present in α-helices, β-strands, and turns. Thornton's group developed several algorithms for identifying ion pairs, hydrogen bonds, and catalytic sites in proteins.5–7 Manavalan and Ponnuswamy 8 proposed the concept of surrounding hydrophobicity to characterize the hydrophobic behavior of amino acid residues in the protein environment. Plaxco et al. 9 analyzed the contacts between amino acid residues and developed the concept of contact order (CO) to relate the folding rates of two-state proteins. Gromiha and Selvaraj 10 considered contacts that are close in space but far away in the sequence and proposed long-range order (LRO) as a parameter for understanding protein-folding rates. This concept was refined by developing multiple contact index, ie, residues having multiple contacts in two- and three-state proteins. 11
Methods are also available to identify binding site residues in protein complexes based on distances between atoms, energetic contributions, and changes in accessible surface area upon binding.12–14 Many standalone programs and online servers (such as DSSP, 15 NACCESS, 16 HYDROPRO, 17 HYDRONMR, 18 GETAREA, 19 SCide, 20 ContPro, 21 CAPTURE, 22 HBPLUS, 23 CALCOM, 24 PSAP, 25 and SBPS 26 ) are available to calculate various structural parameters. For instance, DSSP 15 provides information on the secondary structure and accessible surface area of each amino acid residue in a protein. CALCOM is used to locate residues in the interior and surface based on the distance between the residues and the calculated center of mass of the given protein or peptide chain. 24 Tina et al. 27 developed a server, protein interactions calculator, to calculate the center of mass, hydrogen bond interactions, hydrophobic interactions, aromatic–aromatic interactions, aromatic–sulfur interactions, and cation–π interactions. Kozma et al. 28 developed a server to obtain the contact map for any given protein. Magyar et al. 29 utilized the concept of surrounding hydrophobicity, LRO, stabilization center, and conservation scores to identify the stabilizing residues in protein structures. ExPASy 30 is a collection of tools on various bioinformatic aspects including proteomics, genomics, structural bioinformatics, and systems biology. PDBsum 31 provides pictorial analyses of several structural features of proteins, DNA, and ligands, as well as the interactions between them.
Although a number of structural parameters have been described in the literature and can be calculated using various servers and standalone programs, no single server exists to calculate a diverse set of parameters and provide the output in a standard format. Hence, we have developed a web server, PDBparam (http://www.iitm.ac.in/bioinfo/pdbparam/), to calculate the following four distinct groups of properties: (i) physicochemical properties, (ii) secondary structure propensities, (iii) interresidue interactions, and (iv) identification of binding site residues in protein–DNA/RNA, protein–ligand, and protein–protein complexes. The server and the properties calculated are explained later.
Materials and Methods
A brief description of the properties under the four categories (physicochemical properties, secondary structure propensities, interresidue interactions, and binding site residues in protein complexes) is provided in this section.
Interresidue Interactions
For the past three decades, studies on the mechanism of protein folding and stability have focused on interresidue interactions. 32 Interactions between amino acid residues of the protein and with the surrounding solvent molecules play an important role in the formation of stable secondary structures and a unique tertiary structure for the protein. These interactions are usually noncovalent and include hydrogen bonds, ion pairs, van der Waals interactions, and hydrophobic interactions. In fact, parameters such as CO and LRO show a very strong correlation with the folding rate of small proteins.9,10
Short-, Medium-, and Long-Range Interactions
For a given residue, the surrounding residues within a sphere of 8 Å radius are analyzed in terms of their sequence position. Residues within a distance of two residues from the central residue are considered to contribute to short-range interactions, those within a window between three and four residues to medium-range interactions and those more than four residues apart to long-range interactions.
Number of contacts (8/14 Å, Cα/Cβ atoms)
The contacts between amino acid residues in the crystal structure are computed with cutoffs of 8 and 14 Å using Cα or Cβ atoms, as reported widely in literature. 32
Contact Order
This parameter reflects the relative importance of local and nonlocal contacts to the native structure of a protein. 9 It is defined as

where
Long-range Order
LRO is derived from long-range contacts (contacts between two residues that are close in space and far in the sequence) in the protein structure. 10 It is defined as

where
Total Contact Distance
A new parameter total contact distance was developed by taking the product of CO and LRO. This parameter shows good correlation with the folding rates of proteins. 33
Multiple Contact Index
It considers the distance between amino acid residues in protein structure, residue separation at the sequence level, and the number of residues that have multiple contacts. 11 Multiple contact index has been derived separately for two- and three-state proteins.
Two-state proteins:
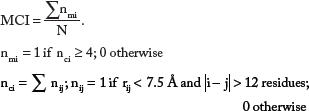
Three-state proteins:

where
Propensities
Propensities indicate the preference of amino acid residues for different secondary structures. The propensities listed in PDBparam are given below.
α-Helical, β-Strand, and Coil Tendencies
The α-helical propensities can be computed by taking into account the frequency of amino acids in these regions.

Frequency of Occurrence in β-Bends
Certain segments in the polypeptide chain help in bringing the distant residues into close proximity during the folding process. For example, β-bends
34
allow hydrogen bonds to form between the C = O group of residue
Criteria to occur in β-bends:
Distance between Cα(
The (
Amino Acid Compositions in Turns
An open turn exists in a protein if the distance between C 1 α to C 4 α carbon atoms is <5.7 Å. 35 Turns are usually present where a strand of β-sheet reverses itself to form the next antiparallel strand or keep the helices, β-sheets, and random coils in a compact globular form and are thus used to predict protein structure.
Normalized Frequency of Helix
Helical regions are divided into three zones 35 : the first three residues represent the N-helix, the last three represent the C-helix, and the residues in the middle represent the M-helix. The amino acid frequency in each helical zone divided by the total frequency (in the entire protein) constitutes normalized frequency.
Propensity to Form Multiple Contact Index
The frequency of occurrence of amino acid residues that form multiple contacts (

where
Amino Acid Composition in High B-Value Regions
Temperature factors (ie,
Physicochemical properties of proteins
Center of mass
The center of mass can be used to define constraints in predicting protein tertiary structures to assess the global shape of the protein partners in protein–protein complexes and to measure their distance.
24
It is given by

Radius of gyration
The radius of gyration describes the compactness of the protein. It is calculated as follows:
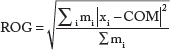
Surrounding hydrophobicity
The sum of hydrophobic indices assigned to the residues that appear within a distance of 8 Å from the central residue
8
can be used to characterize the hydrophobic behavior of each amino acid residue in the protein environment. It is defined as

Gain in surrounding hydrophobicity of a residue
For a given amino acid, the increase in surrounding hydrophobicity as the protein transitions from its unfolded state to its native (ie, folded) state represents the enrichment in the hydrophobic property of that residue. To compute the gain in surrounding hydrophobicity 39 for each residue in the protein molecule, it is assumed that the fully extended chain conformation is the unfolded reference state.
Surrounding hydrophobicity in the unfolded

The average gain ratio in surrounding hydrophobicity is given by

Surface hydrophobicity
This is computed from the protein crystal structure by considering the hydrophobic contribution of exposed amino acid residues. Surface hydrophobicity
38
is given by

Hydrophobic accessible area
It is calculated as the solvent accessible surface area of the hydrophobic residues on the protein surface. 40 We considered Ala, Val, Leu, Ile, Met, Phe, and Pro as the hydrophobic residues to calculate the hydrophobic accessible area.
Accessible surface area for the native protein
The accessible surface area (ASA) for the native protein is calculated as the sum of the accessible surface area of each residue present in the protein, which is obtained from DSSP. 15
Buriedness
The buriedness 2 of each residue is calculated as the ratio of number of residues in the interior of the protein and the total number of residues in the protein.
Mean area buried on transfer
The mean area buried on transfer 41 is given by difference in the accessible area in the unfolded and folded states of the protein.

where
Mean fractional area loss
During the process of folding, the nonpolar residues avoid contact with solvent molecules and are buried inside the protein. The area lost when a residue is buried is proportional to its hydrophobic contribution. This is termed as solvent accessible reduction ratio
41
or mean fractional area loss, denoted as <

where
Normalized flexibility parameters (B-values)
This parameter can be computed from the temperature factors extracted from the PDB for the N, Cα, C, and O atoms. Based on the deviation of
Noncovalent interactions
Several interactions (hydrophobic, hydrogen bond, ionic, aromatic, cation–Π, and disulfide bonds) have been described in terms of the amino acid residues involved and the distance between two specific amino acid residues. The details of the amino acid residues in each interaction along with the distances 27 are given in Table 1.
Distance criteria for noncovalent interactions and disulfide bonds.

Hydrophobic-free energy
The hydrophobic-free energy 43 is expressed as

where
The solvent accessible surface areas of all the atoms in the folded state were computed using the program NACCESS.
16
The extended state ASA of the atom was obtained from literature. They are in the form of a Gly–X–Gly (where × is the amino acid) sequence in a typical extended conformation. σ
Free energy due to disulfide interactions
The free energy due to disulfide interactions is calculated using the formula:

where
Hydrogen bond interactions
It is classified into the following three main categories: main chain–main chain, main chain–side chain, and side chain–side chain interactions. These interactions are calculated using HBPLUS, 23 a hydrogen bond calculation program.
Identification of binding sites in protein–DNA/RNA and protein–protein complexes
Protein–DNA interactions play a key role in many vital processes, including regulation of gene expression, DNA replication and repair, and packaging. The binding sites for a protein–DNA/RNA complex can be identified using the following distance criteria 12 : an amino acid residue within a protein is designated as a binding site residue if its side chain or backbone atoms are within a cutoff distance (eg, 3.5 Å) from any atom in DNA/RNA.44–46 The binding sites for protein–protein complexes were also computed using the distance criteria between different chains present in the protein.
Server Description and Implementation
The PDBparam server can calculate more than 50 parameters from the three-dimensional structure of a protein. Each parameter has been treated as a separate module, and the script has been written using perl. The perl-CGI scripts are used to render the HTML web pages. The PDBparam server works with the PDB file as input and provides the computed results in a single output page. The output can be downloaded as a PDF file. The results for all the parameters were cross-checked manually with several structures of proteins and their complexes. Furthermore, the documentation has been provided for all the parameters listed in PDBparam on the website. It is linked with other online tools available in the literature. The utility of the server is described with a few examples.
Steps:
Enter the PDB code and chain (optional; case sensitive); eg, PDB code: 6CRO.
Check “identification of binding site” and submit.
In the new page, check protein–DNA/RNA.
Give the distance (default cutoff is 3.5 Å).
Click on submit.
Figure 1 shows the relevant items to be checked, the required information, and the output. The output contains information on the residue name, residue number, atom name, and chain name of both protein and DNA and the distance between the atoms. These residues are identified as binding sites. We have also provided options to display the structure of the complex, highlighting the binding site residues.
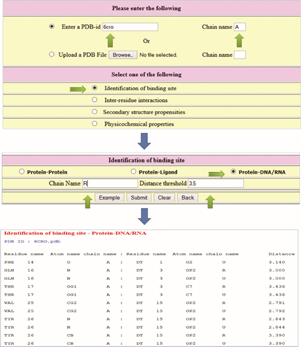
Steps to identify the binding sites in a protein–DNA complex.
Steps:
Enter the PDB code and chain (optional; case sensitive). Check “interresidue interactions” and submit. In the new page, check “contact order and number of contacts (8 Å, CA atoms)”. Click on submit.
Figure 2 shows the relevant items for computing the CO and number of contacts and the output. The output displays the CO for the protein and the number of contacts for all the residues with residue name and number. The contacting residues are also shown in the output.

Example to compute the contact order of a protein and the number of contacts for all the amino acid residues in a protein.
Availability of PDBparam
PDBparam is freely available at http://www.iitm.ac.in/bioinfo/pdbparam.
Applications
PDBparam computes various structure-based parameters on interresidue interactions, amino acid propensities, physicochemical properties, and binding sites. This information can be used to understand the structure and functions of proteins and their complexes. The contacts between amino acid residues in protein structures provide data on the location of amino acid residues and preferred contacts in the protein environment, which can be used to comprehend protein folding and predict protein structures. 32 The topological parameters, such as CO, LRO, total contact distance, and multiple contact distance, are helpful in understanding protein-folding rates and folding kinetics.9–11 Specific physicochemical interactions between amino acid residues in protein structures, such as cation–π, aromatic clusters, and hydrogen bonds, reveal the importance of these interactions inproteinstability. 27 The combination of secondary structure and solvent accessibility is useful in identifying functionally important residues in proteins.15,16 Furthermore, the identification of binding sites in protein–protein, protein–nucleic acid, and protein–ligand complexes can be effectively used to compute the binding propensity and affinity and understand the recognition mechanism of protein complexes.46–51
PDBparam can be used to compute important parameters for any specific protein, providing deep insights into its structure–function relationship. It can also be used for large-scale analysis of different types of proteins to explore potential interactions and contacts, which will provide insights on the similarities and differences crucial to understanding the function.
Conclusion
The PDBparam server can calculate more than 50 parameters from the three-dimensional structure of a protein, classified into the following four categories: physicochemical properties, interresidue interactions, secondary structure propensities, and identification of binding sites in protein–DNA/RNA and protein–protein complexes. All the parameters have been coded using perl. Furthermore, perl-CGI scripts are used to render the HTML web pages. Detailed documentation for the protein properties and links of other available web servers related to such properties are provided, in order to enhance the user's ease of access.
Author Contributions
Conceived and designed the study: MMG, DV. Web server development: AA, AMT, RN. Discussions: AA, AMT, RN, SJ, DV, MMG. Wrote the first draft of the article: AA, MMG. Contributed to the writing of the article: AMT, RN, SJ, DV. All the authors reviewed and approved the final article.
Footnotes
Acknowledgment
We thank the Bioinformatics Facility, Department of Biotechnology, and IIT Madras for computational facilities.