
Abstract
Chaetognatha is a minor phylum, comprising transparent marine invertebrates varying in size from 0.5 to 12 cm. The exact phylogenetic position of Chaetognatha in Metazoa has not been deciphered as some embryological characteristics place chaetognaths among deuterostomes and some morphological characteristics place these among protostomes. In this study, the major factors that drive synonymous codon usage bias (SCUB) in the mitogenomes of representative species of Chaetognatha and chosen species of other closely related phyla were analyzed. Spearman's rank correlation analyses of nucleotide contents suggested that mutational pressure and selection were acting in all examined mitogenomes but with varying intensities. The quantification of SCUB using effective number of codons vs. GC composition at the third codon position (GC3) plot suggested that mutational pressure due to GC compositional constraints might be one of the major influencing forces driving the SCUB in all chaetognaths except
Introduction
Most of the amino acids are encoded by more than one codon in the protein-coding genes (PCGs), and those codons are termed as synonymous codons. Synonymous codons have been observed to be used at unequal frequencies in various genomes. Synonymous codon usage (SCU) is species specific.
1
In certain genomes, a subset of codons is used most frequently to encode a given amino acid and this bias in codon usage exists in a wide variety of organisms, from prokaryotes to unicellular and multicellular eukaryotes.
1
Mutational pressure and natural selection have been identified as the two major evolutionary forces that contribute to bias in SCU (SCUB).2–5 SCU preferences can be attributed to a variety of factors that vary from one species to another.6,7 In unicellular prokaryotic organisms such as
Though mutational pressure and natural selection have been identified as two major evolutionary forces that drive SCUB, in certain eukaryotes, SCUB and usage of optimal codons are often influenced by certain selective forces, 15 which are unknown. Investigation of the pattern of codon usage and the various factors influencing its diversification form the basis for understanding the evolution of genomes and the habitat adaptation of living organisms.16–18 Nuclear genomes of various organisms have been exclusively analyzed to detect trends associated with codon usage.19,20 However, mitochondrial genomes have been less studied so far, and only very few studies have been carried out on mitogenomes.21,22
Chaetognatha is a minor phylum, comprising transparent marine invertebrates varying in size from 0.5 to 12 cm.23,24 They feed on small animals such as copepods and immature fish. 23 Because chaetognaths are very ancient, only little information is available about their evolutionary origin. 25 The exact phylogenetic position of Chaetognatha in Metazoa is not clearly understood as some embryological characteristics place chaetognaths among deuterostomes and some morphological characteristics place them among protostomes. 26 But, some studies have supported the inclusion of chaetognaths among protostomes.23,24 The inclusion of Chaetognatha at the appropriate position in the bilaterian lineage has been regarded as one of the most important issues in metazoan phylogeny. 27
Considering these facts, this study has been designed with the aim of analyzing the various trends associated with SCU in the mitogenomes of all sequenced chaetognaths and other closely related phyla. In this study, evolution of SCUB in the representative species of protostomes and deuterostomes has been extensively studied and compared with that of chaetognaths. The study would certainly help to get insight into the evolution of SCUB of chaetognaths and other chosen species belonging to the bilaterian lineage. The main objective of this study was to analyze the major factors that dictate SCUB in chaetognaths as well as representative species chosen from among protostomes and deuterostomes. This comparative study reveals the closeness of chaetognaths to protostomes in terms of SCUB.
Materials and Methods
Sequence Data
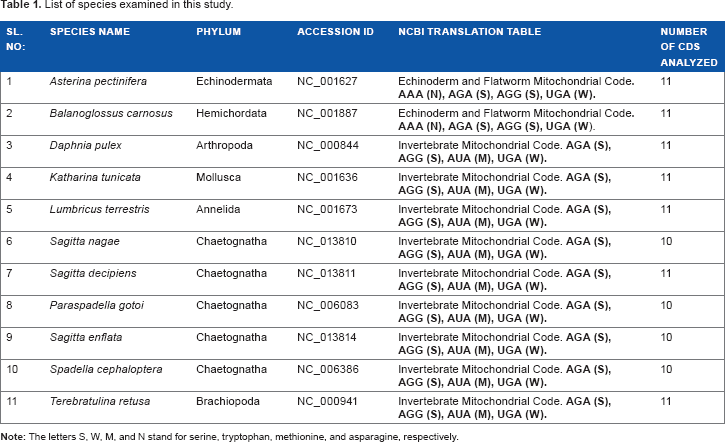
Complete mitochondrial genomes of five representative species from the minor phylum Chaetognatha, as well as for one representative species each from Hemichordata, Echinodermata, Arthropoda, Annelida, Mollusca, and Brachiopoda, were retrieved from the National Center for Biotechnology Information (NCBI) (Table 1). The complete coding sequences (CDSs) were examined for the presence of proper initiation and termination codons at the beginning and at the end of the sequences in order to ensure integrity. The final data set contains only sequences having more than 300 nucleotides to avoid sampling errors leading to statistical fluctuations, as suggested in a recent study. 22 The number of CDSs chosen for this study is provided in Table 1.
List of species examined in this study.
Measures of SCU
Relative SCU
Relative SCU (RSCU) for each informative codon was calculated for the normalization of SCU by avoiding the influence of amino acid composition. The RSCU values are considered to be independent of gene length. 22 RSCU values were obtained as the ratio of the observed frequency of a codon to the expected frequency of that particular codon provided all synonymous codons for the same amino acid are used equally. 8 If the RSCU value is greater than 1, it indicates a bias toward that particular codon, and if the RSCU value is close to 0, it indicates lack of bias. 17
Plot of Effective Number of Codons vs GC3
The extent of SCUB is often quantified using the effective number of codons (ENC), which is independent of gene length and is an indicator of the level of gene expression. 28 ENC is independent of constraints of amino acid composition 22 and takes values ranging from 20 to 61. 28 If the bias is extreme, the ENC would be 20, ie, only one codon would be used for coding one amino acid, and the ENC would be 61 if all synonymous codons for a particular amino acid are used equally, ie, there is no bias at all. 28 If the ENC value of a gene is less than 35, bias is regarded to be high, and if the ENC value is above 50, bias is regarded to be low. 29 The overall and local GC compositions (GC content at the three codon positions) were calculated after excluding the stop codons. The expected ENC values were computed using GC3 (presence of a guanine or a cytosine at the third codon position) under the assumption that no selection exists. 28 In this study, ENC was plotted against GC3 to obtain a visual display of the influence of major evolutionary forces, such as mutational pressure and natural selection, on SCUB by the scattering of genes along the expected ENC curve. 28 The ENC values and GC3 values for all PCGs of the selected mitogenomes were calculated and the ENC-vs-GC3 plot was developed.
Neutrality Plot
Cumulative GC content at the first and second codon positions (GC12) and the GC composition at the third codon position (GC3) were used to derive the neutrality plot to understand the influence of GC composition in the shaping of codon usage. 17 The influence of mutational pressure on SCUB is low, if the slope of the regression line is close to 0 and it is high if the slope is close to 1.17,30
Parity Rule 2 Plot
Parity rule 2 bias plot (PR2 bias plot) was developed in order to detect the biases for A ≈ T and G ≈ C within a DNA strand (Second parity rule of Chargaff). 31 In the PR2 bias plot, only the third codon positions in the fourfold degenerate amino acids were plotted. 31
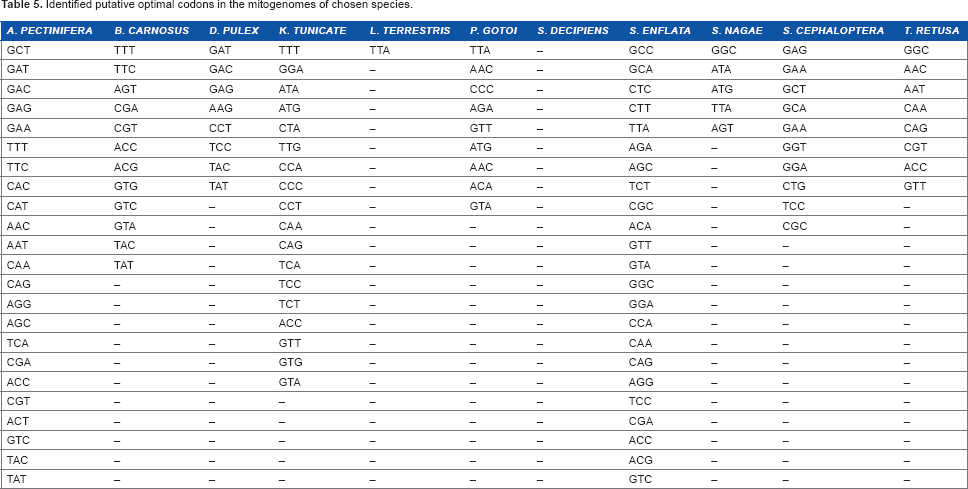
Identification of Putative Optimal Codons
The putative optimal codons were identified using chi-square test for a 2 × 2 matrix having one degree of freedom.
32
Two data sets for a 2 × 2 matrix were developed by selecting 10% of the genes that were grouped on the left and the right extremes of the first axis of the correspondence analysis (COA).33,34 Observed frequencies of codons in the two data sets are given in the first row and the total number of synonymous alternatives of that given codon is given in the second row of the 2 × 2 matrix.
30
Level of significance was measured at
Codon Adaptation Index
The codon adaptation index (CAI) 35 was calculated to understand the magnitude of bias in the analyzed mitogenomes toward a subset of referred codons in highly expressed genes. The CAI value ranges from 0 to 1 and a value close to 1 indicates higher codon usage bias and level of gene expression. 35
Correspondence Analysis
COA was performed on the RSCU (COA–RSCU) for identifying variations in SCU by avoiding the influence of amino acid composition. 29 COA partitions the variations along certain numbers of orthogonal axes (the number of orthogonal axes depends upon the total number of synonymous codons excluding stop codons). 36 The first axis accounts for the majority of the variation, with each subsequent axis accounting for diminishing amount of variance. 36 COA considers each CDS as a 59-dimensional vector, and each dimension is defined by the RSCU value of a particular informative codon. 37
Cluster Analysis
Cluster analysis on the RSCU values was performed to show the association between codon bias and other factors. 29 In the cluster analysis, a matrix in which the rows and columns correspond to chosen species and pooled RSCU values of synonymous codons, respectively, was generated. 29 Unweighted pair-group average clustering grouped the selected species based on the variations in the RSCU values, and the distances are in Euclidean distance. 29
Statistical Analysis and Software Implementation
Nonparametric Spearman's rank correlation method was used for all correlation analyses between various codon usage indexes and other important parameters as it makes no assumptions about the probability relation between the two variables.30,36 PAST software version 2.12 was used for Spearman's rank correlation analysis and cluster analysis. 38 ENC, aromaticity, and Grand Average of Hydropathy (GRAVY) scores were estimated using CodonW (online version: http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms:codonw). 39 Overall and local nucleotide compositions were calculated using MEGA 5.2.2. 40 RSCU values were calculated using DAMBE version 5.3.31. 41 CAI values were computed using ACUA 1.0 42
Results
Compositional Properties Influence Codon Usage in the Analyzed Mitogenomes
Complex correlations were noticed between overall and silent base compositions in the examined mitogenomes (Table 2). Significant positive correlations were observed between T and GC3, C and T3 in
Spearman's rank correlation analysis between overall and silent base contents.

Analysis of RSCU
Overall and strand-specific codon usages were analyzed (Tables 3 and 4). Significant strand-specific codon usage bias was not observed in any of the genomes (Table 4). In
Overall relative synonymous codon usage in the mitogenomes of analyzed genomes.

Strand-specific relative synonymous codon usage values.

Identified putative optimal codons in the mitogenomes of chosen species.
Quantification of SCUB
In

ENC vs. GC3 plots for the quantification of SCUB. The SCUB of genes lying on or close to the expected GC3 is dictated by GC compositional constraints.

Neutrality plot. If correlation exists between GC3 and GC12, GC compositional constraints may have profound influence in shaping SCUB. If no correlation exists between GC3 and GC12, selection may have significant role in framing SCUB.
To analyze whether the SCUB is restricted in extremely biased PCGs, the relationship between purines (A and G) and pyrimidines (C and T) in four-codon amino acid families was analyzed using the PR2 bias plot (Fig. 3). Complex associations between purines and pyrimidines were observed in Chaetognatha and the other examined genomes. The analysis of PR2 bias plot revealed that A and T contents were used more proportionally than G and C contents. In addition, significant compositional differences between C and G and between A and T contents were found in most of the PCGs.

Parity rule 2 bias plot. This plot reveals the proportionate usage of A and T contents in comparison with that of G and C contents.
Factors Influencing Codon Usage Bias
COA–RSCU was carried out to identify the major factors influencing the SCUB (Fig. 4). Correlation between codon usage indexes and COA axes was analyzed using Spearman's rank correlation method (Table 6). Axis 1 and Axis 2 accounted for the majority of the total variations. Thus, Axis 1 could be considered to be a single major explanatory axis as it accounted for more than 40% variations in all chosen species. In

Correspondence analysis. Axis 1 accounts for the majority of variations. COA–RSCU plot reveals various factors that are responsible for the variations in RSCU values.
Spearman's aqrank correlation analysis between various codon usage indexes and major axes of COA.

P ≤ 0.05;
P ≤ 0.01.
Cluster analysis yielded two clusters, ie, one upper cluster (major) and one lower cluster (minor), based on the variation in RSCU values (Fig. 5). The upper cluster included all chosen species from Chaetognatha, Mollusca, and Arthropoda, and the lower minor cluster included the species chosen from Annelida, Echinodermata, Brachiopoda, and Hemichordata. This grouping suggested that variations of RSCU values in the PCGs of mitogenomes in the chaetognaths were more similar to those of the chosen species of protostomes than those of deuterostomes. The results suggest that mutational pressure, natural selection, and some other unknown selective forces were acting at varying intensities (species specific) in the examined mitogenomes (Fig. 6). In general, terms such as strong, moderate, and weak are used in Figure 6 to express the idea that in any selected mitogenome, the intensity of mutational pressure and selection pressure vary significantly. In this study, this factor was assessed by using the following strategy.

Cluster analysis. All examined species were grouped into two clusters based on the variations in RSCU values. All chaetognaths were grouped with species belonging to protostomes.

Identified major forces driving SCUB in mitogenomes of chaetognaths and other closely related phyla. The intensity of these forces vary and terms such as strong, moderate, and weak are used to express it.
Among the following five conditions,
no significant correlation between GC3 and GC12;
significant correlation between Axis 1 COA, with the indexes showing the level of gene expression;
positive correlation between heterogeneous nucleotide contents;
negative correlation between homogeneous nucleotide contents; and
significant correlation between Axis 1 COA and any of the silent base contents,
if any three conditions are met, then it is considered that strong selection pressure acts on such genomes. If two conditions are met, then the term “moderate” is used to express the intensity of selection, and if only one or no condition is met, the term “weak” is used. The intensity of mutational pressure was also assessed by using the above strategy, albeit vice versa. If both mutational and selection pressures are strong, then the influence of the unknown selective forces on SCUB was reckoned as weak.
Discussion
Natural selection and mutational pressure are regarded as the two major forces behind the species-specific SCU.17,36 Analysis of codon usage revealed that influence of selection or mutational pressure was not absolute in representative species of Chaetognatha. In
We confirmed that trends associated with codon usage in the representative species of the minor phylum Chaetognatha were not uniform and absolute, and these findings were supported by the genome theory that codon usage is species specific. 44 In addition, we suggest that SCU variation in the mitogenomes of chaetognaths was not completely dictated by GC3 composition and selection pressure. No correlation between GC12 and GC3 in the mitogenomes of selected chaetognaths indicated that mutational pressure due to GC compositional constraints was apparently less influential in framing the SCUB in the mitogenomes of these species. Our analyses showed the presence of certain other unknown selective forces in shaping codon usage across PCGs in the mitogenomes of species belonging to Chaetognatha.
In general, the unknown selective forces act at the mRNA level. 15 The mRNA sequences contain both information (coding sequences) required for synthesizing protein as well as information (motifs) required for regulating the expression. 15 It is common knowledge that most of the motifs are mRNA secondary structures. Though the functional significance of these mRNA secondary structures are not fully understood, 15 a few studies have reported that these structures interfere with the process of translation44,45 and the stability of such structures is higher than expected.46–48 If highly stable mRNA secondary structures interfere with the process of translation, it is obvious that selective forces will act against such stable secondary structures. 15 But, no foolproof method has been developed for predicting the structure of mRNA molecules as the structures predicted using thermodynamic and comparative methods possess several drawbacks. 15 These methods predict the structures that are best approximations of the mRNA secondary structures present inside the cell and, hence, in general, it is appropriate to use the term “unknown selective forces” to express “seemingly profound influences” that are not explainable on the basis of natural selection and mutational pressure. 15 Note that even extremely biased genes use nonoptimal codons, as usage of optimal codons vary according to the influence of alternative selective forces, depending upon the strength and direction of translational selection. 15 Because the accuracy of the mRNA structures predicted using bioinformatic tools is still questionable, it has been suggested that there may exist selective forces 15 that are yet to be known.
In
Cluster analysis based on RSCU values revealed that RSCU variations in the PCGs of the mitogenomes of chaetognaths are more comparable with those in protostomes as all chosen species of Chaetognatha, Mollusca, and Arthropoda formed one major cluster (upper cluster).
The putative optimal codons identified in the mitogenomes of the chosen species of Chaetognatha and those of other phyla could be correlated with the gene expression levels. 30 The presence of putative optimal codons could be suggested as a marker for genes having unknown expression levels. 30 Because the frequency of optimal codons was found to vary in all representative species of Chaetognatha, optimal codon usage was not considered to be conserved among these species. 49 Putative optimal codons are of paramount significance in enhancing heterologous gene expression and, moreover, the a software named Visual Gene Developer improves the expression levels of a synthetic gene by adopting a codon optimization strategy.50,51 Hence, the identification of putative optimal codons in chaetognaths may help in studies relating to transcriptional control of gene expression as chaetognaths are regarded as good model systems for comparative genomics to study the evolution of animal genomes. 52
This study was able to identify the trends associated with SCU in the representative species of the minor phylum Chaetognatha and other closely related phyla, such as Hemichordata, Echinodermata, Brachiopoda (deuterostomes), Mollusca, Annelida, and Arthropoda (protostomes). From the results, we understand that some forces other than mutational and selection pressures act on the PCGs in the mitogenomes of all chosen species. The high degree of complexity in the process of speciation of chaetognaths 53 might have invited certain other forces that can interfere with the SCUB. We conclude that the identification of these unknown selective forces would certainly help get insight into the evolution of mitogenomes in chaetognaths.
Author Contributions
Designed the work: RRN, VRD. Analyzed the data: SK, RRN, VRD. Drafted the manuscript: RRN, VRD. Executed the work according to the designed methodology and collected data: SK, VN, US, NSSK. All the authors reviewed and approved the final draft of the manuscript.