
Abstract
The evolution of bias in synonymous codon usage in chosen monkeypox viral genomes and the factors influencing its diversification have not been reported so far. In this study, various trends associated with synonymous codon usage in chosen monkeypox viral genomes were investigated, and the results are reported. Identification of factors that influence codon usage in chosen monkeypox viral genomes was done using various codon usage indices, such as the relative synonymous codon usage, the effective number of codons, and the codon adaptation index. The Spearman rank correlation analysis and a correspondence analysis were used for correlating various factors with codon usage. The results revealed that mutational pressure due to compositional constraints, gene expression level, and selection at the codon level for utilization of putative optimal codons are major factors influencing synonymous codon usage bias in monkeypox viral genomes. A cluster analysis of relative synonymous codon usage values revealed a grouping of more virulent strains as one major cluster (Central African strains) and a grouping of less virulent strains (West African strains) as another major cluster, indicating a relationship between virulence and synonymous codon usage bias. This study concluded that a balance between the mutational pressure acting at the base composition level and the selection pressure acting at the amino acid level frames synonymous codon usage bias in the chosen monkeypox viruses. The natural selection from the host does not seem to have influenced the synonymous codon usage bias in the analyzed monkeypox viral genomes.
Keywords
Introduction
Molecular evolution is a broad term reflecting changes in various genomic parameters due to alterations in the nucleotide and the dinucleotide compositions that lead to an accumulation of mutations over time. 1 Because the genetic code is degenerate, more than one codon can encode a particular amino acid; however, the usage of these “synonymous codons” for a given amino acid is not uniform. 2 In a given amino acid, a subset of codons may be used more frequently than others are, and such a subset is referred to as “preferred codons.” 3 Synonymous codon usage bias (SCUB) is species specific and varies within and between genomes. 4 This nonuniform usage of synonymous codons (ie, SCUB) can be significant in highly expressed genes. 5 Thus, an understanding of SCUB is critical as it reveals the various forces that frame genomic evolution. 6
The mutational pressure, which is due to base compositional constraints, and the selection pressure, which increases the translational speed and accuracy, have been identified as 2 important forces causing SCUB in various lineages, such as plants, mammals, macro-invertebrates, bacteria, fungi, and viruses.7–11 Selection pressure favors codons having abundant transfer RNAs, particularly in highly expressed genes.12–15 Furthermore, synonymous codon choices for protein formation have been found to affect secondary structure and protein folding, 16 and messenger RNAs (mRNAs) and protein structures have been found to cause selection pressure.17–19 For instance, a significant species-specific correlation was noticed between the usage of AAC (asparagine) and the C-terminal regions of β-sheet segments in Escherichia coli as selection for translational efficiency favors downstream asparagine (AAC) residues that are essential for the formation of the β-sheet. 19 Similarly, a significant correlation was found to exist between GAU (aspartic acid) and the N-termini of α-helices in humans as selection acts on co-translational protein folding in eukaryotes. 19 In an another important study, selection on synonymous codon usage (SCU) facilitated the optimization of the characteristics of mRNA secondary structures as a specific codon usage pattern was observed in the nucleotide sequence of repetitive units of silk fibroin mRNA. 17 However, in another study, the mutational pressure was found to frame the overall nucleotide composition in genomes through GC « » AT changes, 20 and intrinsic bias in dinucleotide frequencies may have had an influence on SCUB 6 as such bias can be extreme. 20 For instance, the CpG (C-phosphate-G) content is underrepresented in many vertebrates owing to the methylation of cytosine residues, 21 and the TpA (T-phosphate-A) content is restricted in many organisms due to the susceptibility of uracil in UpA to RNase 22 and low thermal stability. 23
The quantification of SCUB and the identification of its causative factors in zoonotic viral genomes are crucial in understanding viral evolution and ecology. 6 Detailed analyses of trends and SCUB-associated factors are essential if the mechanisms of viral infection and immune response are to be revealed. 20 Greater emphasis on understanding the various factors contributing to codon usage patterns is, therefore, more important than merely understanding viral SCUB.24–28 The survival, fitness, and evolution of viruses depend strongly on SCUB coactions between viruses and hosts because replication and translation of viral genomes are host associated. 20 Few studies have been undertaken to reveal the major forces and trends associated with viral DNA SCUB.20,29,30 Substantial differences between the SCUB in a virus and that in its host will have an effect on viral replication and protein synthesis, 31 as evidenced in human papillomaviruses. 32
Monkeypox viruses (MPXVs) belong to the genus Orthopoxvirus of the family Poxviridae. 33 The family Poxviridae consists of large double-stranded DNA viruses capable of replicating in the cytoplasm of vertebrate and invertebrate cells.33,34 Monkeypox viruses cause human diseases similar to the eradicated smallpox caused by the variola virus (VAR). 33 By 1977, smallpox was reported to have been eradicated and vaccination was stopped. 35 As a result, closely related zoonotic viruses such as MPXVs infected unvaccinated human populations and caused a fatal illness (human monkeypox), but with a very low human-to-human transmission rate.36,37 Although human monkeypox is clinically similar to smallpox, regarding the case fatality rates (CFRs), smallpox was reported to be severe than human monkeypox, with the former having a CFR of 30% 38 and the latter a CFR of 10%. 36 A recent outbreak investigation conducted in the Bokungu Health Zone of the Democratic Republic of the Congo (DRC) from July 1 to December 8, 2013, revealed a 600-fold increase in the number of human monkeypox cases. 39
Rodents are the major animal reservoirs for MPXVs.40–42 The viral transmission to humans takes place through direct contact with animals. 43 Wounds in the skin are the major route through which infection happens while handling infected animals. 41 In some cases, respiratory transmission from animal to human and then from human to human has occurred.41,44 The incubation period is 10 to 14 days. 43 After the incubation period, the prodromal period lasts for 2 days, and in this phase, the infected individual may experience fever, chills, malaise, headache, backache, sore throat, shortness of breath, and swollen lymph nodes.45,46 A clinical feature that can be used to differentiate between human monkeypox and human smallpox infections is the presence of enlarged lymph nodes in the submandibular, cervical, or inguinal regions in the former. 35 The infected individual becomes most contagious subsequent to the development of a progressive maculopapular rash (0.2-1.0 cm) after the prodromal period.45,46 The spread of the lesions over the body follows a centrifugal pattern, and in certain cases, dyspigmented scars may develop from the lesions. 43 In general, during a 2- to 4-week time period, the lesions over the body progressively undergo several changes from macules to papules, vesicles, and pustules, followed by scabbing and desquamation.35,43
Human monkeypox is endemic to the DRC, and infections take place throughout the Congo Basin. 39 Different isolates of MPXVs from West Africa and the Congo Basin have been proven to be genetically distinct, and substantial differences in virulence between them have been reported. 47 For instance, MPXV-ZAI-V79 isolated from the Congo Basin is thought to be more virulent than MPXV-COP-58 isolated from West Africa 47 as no mortalities were reported during the West African isolate MPXV outbreaks in the United States in 2003. 47 However, high virulence (>90%) and fatalities have been reported in the Congo Basin, and D10L, D14L, B10R, B14R, and B19R have been identified as possible candidate loci for virulence. 47 Although genetic analyses revealed that MPXVs are not the immediate ancestors of the VAR because considerable differences were found between MPXVs and the VAR in the terminal genomic regions encoding virulence and host range factors, the possibility of an MPXV evolving into a highly virulent VAR-like virus with significant human-to-human transmission rates cannot be ignored. 37
In this study, extensive analyses of SCUBs in 13 representative MPXV genomes isolated from different African regions were conducted to unravel patterns and factors associated with MPXV diversification. The size of the double-stranded DNA genome of an MPXV is ≈200 kb, comprising ≈190 nonoverlapping open reading frames (ORFs) that contain ≥180 nucleotides. 48 A typical monkeypox genome contains a central conserved region (≈560 00 to 120 000 nucleotides long), with variable regions to the left and the right, as well as an inverted terminal region (ITR) with tandem repeats. 33 The central conserved region contains genes with the codes for the replication machinery. 48 The ITR in the MPXV genome represents a global repeat49,50 and accounts for almost 1% of the total genome size.50,51 At least 4 ORFs are included in the ITR of the MPXV genomes.52,53 The ORFs in the ITR take part in the virus-host interactions.48,54
As differences in virulence regarding location have been reported, 47 an objective of this study was to reveal associations between virulence and various trends associated with SCUB in MPXV genomes. The results of this research should contribute to an understanding of the coaction between the genome-wide neutral mutational and selection pressures, which, in turn, increases our understanding of viral DNA evolution, as well as the interactions between the viruses and their hosts. Most importantly, the results of SCUB analyses of viral genomes should have important applications in studies related to the genetic engineering of viral genome sequences. 20
Materials and Methods
Sequence data
The complete genomes of 13 representative MPXVs (Table 1) were retrieved from the National Center for Biotechnology Information. Details such as accession numbers, the region of isolation, the number of coding sequences (CDSs) selected, and the sizes of the genomes were also provided (Table 1). The integrity of full-length coding sequences without introns was confirmed by checking for the presence of proper initiation and termination codons. 55 To avoid sampling errors and stochastic variations, we chose CDSs having more than 300 nucleotides for analysis (Table 1). 8 Information regarding the ITRs of the MPXV genomes was obtained from GenBank, and for the calculation of the codon usage in an ITR, the orientation was changed in such a way as to maintain the corresponding amino acid sequences intact and thereby avoid any miscalculation of the codon usage.
Details of examined monkeypox virus strains.

Measures of SCU
The effective number of codons (ENC) is a commonly employed index for measuring SCUB independently of the length of the CDS. 56 The ENC values vary from 20 to 61. In any given gene, if only one codon is used to encode one particular amino acid, the ENC value will be 20 (extreme SCUB). If all synonymous codons of a particular amino acid are used equally, the ENC value will be 61 (almost no SCUB). The compositions of the G and the C nucleotides were calculated for the first, second, and third codon positions. Expected ENC values were calculated using the GC3 (GC composition at the third codon position) values. 56 An ENC versus GC3 plot can be used to distinguish between the 2 major evolutionary forces, the mutational pressure and the translational selection, for the observed SCU patterns by displaying gene groupings along the expected ENC curve. This is true because these 2 major evolutionary forces are the ones that contribute to SCUB. Even though, in some cases, genetic drift can be considered as a factor shaping codon usage; the ENC versus GC3 plot will only give an indication of the influences of the mutational pressure and the selection pressure. In this research, ENC values were calculated according to the following equation 56 :

where F2, F3, F4, and F6 are the average homozygosity values for 4 different synonymous family types and were estimated using the codon frequencies squared. The average homozygosity for each amino acid was calculated according to the following equation 56 :
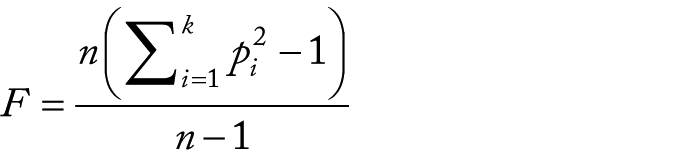
where k is the number of alleles squared. The expected ENC versus GC3 curve was plotted using GC3 values ranging from 0% to 100% in intervals of 10% and their corresponding expected ENC values, which, under no selection, can be calculated using the following equation 56 :

where s = GC3.
The relative SCU (RSCU), which is the ratio of the observed codon frequency to the expected codon frequency, provided all synonymous codons of that particular amino acid have uniform usage, is another important index for measuring SCUB.3,12 The RSCU values greater than 1 denote codons used more frequently than their synonymous counterparts, whereas the RSCU values less than 1 represent codons used less frequently; codons with an RSCU value of 1 denote no bias. 3
The codon adaptation index (CAI) assesses the significance of selection in shaping the observed patterns of the SCU of a gene 5 using a reference set of highly expressed genes from a particular species. The CAI indicates the level of gene expression5,10,11 by calculating a score for each gene. The CAI values from 0.75 to 1.0 indicate a high level of gene expression. 5 Although the CAI is independent of gene length, the CAI of short genes may be affected by sampling bias. 5 We used the Homo sapiens general codon usage table as a reference set because the CAI is a good indicator of viral gene adaptation to the host. 5
Protein hydrophobicity and aromaticity (ie, frequency of aromatic amino acids such as Phe, Trp, and Tyr) were calculated. 57 A correspondence analysis of RSCU (COA-RSCU) has been generally adopted to identify intragenomic variations while avoiding the influence of the amino acid’s composition.8,11 In a COA-RSCU, each CDS is represented as a 59-dimensional vector, 58 wherein each dimension corresponds to the RSCU value of a particular codon. 58 The COA-RSCU partitions the total variation in codon usage across 59 orthogonal axes with 41 degrees of freedom. 8 The first axis of the COA-RSCU (axis 1) accounts for most of variations, whereas subsequent axes capture decreasing amounts of variance. 8
Putative optimal codons were identified by applying the χ2 test to a 2 × 2 matrix having 1 degree of freedom. We chose 10% of the genes lying on the left and the right extremes of axis 1 of the COA-RSCU to form 2 data sets as axis 1 of the COA-RSCU accounts for most of the variations in the RSCU. The first row of this matrix contains the observed codon frequencies from the 2 data sets, whereas the second row contains the total number of synonymous alternatives of that particular codon. 8 Codons whose frequencies of usage were significantly higher (P < .05) in one data set than in the other data set were defined as putative optimal codons.
Cluster analysis
A cluster analysis of the RSCU values was performed to reveal the relationship between the SCUB and other factors based on groupings of the codon usage. 7 In the cluster analysis, a 13 × 59 matrix, in which rows and columns corresponded to the 13 MPXV strains and the pooled RSCU values of the 59 codon species, respectively, was generated. Clustering of the MPXVs based on RSCU values was found to have occurred using unweighted pair-group average clustering and Euclidean distances.
Statistical analysis
The nonparametric Spearman rank correlation was adopted for all correlation analyses between the various codon usage indices and the other parameters as it does not hold any assumptions regarding the distribution of underlying data.8,55 The Mann-Whitney 2-sample test was used to analyze the intergenomic differences in the ENC values. PAST software version 2.12 was used for the Spearman rank correlation analysis. 59 CodonW (http://mobyle.pasteur.fr/cgi-bin/portal.py?#forms::codonw) was used to compute the values of the ENC, hydrophobicity, and aromaticity. 60 MEGA version 5.2.2 was used to calculate the compositions of the nucleotides. 61 DAMBE version 5.3.31 was employed to determine the RSCU values, 62 and the CAI values were computed using ACUA 1.0. 63 The level of significance was taken as .05.
Results
Effect of base composition on SCUB
The overall and the wobble base contents were estimated in all 13 examined MPXV genomes. Overall, the AT content was found to be higher than the GC content. Among the individual nucleotide compositions, the A content was higher than the T, G, and C contents and varied by 35.26 ± 0.053; thus, it was overrepresented in the protein-coding genes (PCGs) of all genomes. In all examined genomes, the C content was observed to be the least among all other nucleotide contents and to vary by 15.52 ± 0.025; thus, it was underrepresented in the PCGs of all genomes. Moreover, the GC content was observed to vary by 33.74 ± 0.065 in all genomes. Because the base changes that occur at the third site of synonymous codons for a given amino acid are neutral, the third site of a codon is commonly known as “the silent site.” Interestingly, the T3 content was higher than the contents of other silent bases (A3, G3, and C3) and was found to vary by 38.23 ± 0.082; the GC composition at silent sites (GC3) was found to vary by 29.12 ± 0.080.
A Spearman rank correlation analysis revealed complex correlations between the overall and the silent base compositions, indicating the presence of compositional constraints in all genomes. The existence of positive correlations between homogeneous nucleotide contents and negative correlations between heterogeneous nucleotide contents implies that mutational pressure due to compositional constraints might play a crucial role in shaping the codon usage. 64 In the case of viral genomes, the positively correlated heterogeneous contents and the negatively correlated homogeneous contents indicate natural selection by the host. 24 In this study, significant positive correlations were found between A and A3, T and T3, G and G3, and C and C3. The most heterogeneous base contents were found for significant negative correlations (Table 2). The G3, C3, and GC3 contents were found to have significant positive correlations with the overall GC content. No correlations were observed between G3 and C, T3 and A, and vice versa. These noncorrelations did not reveal any SCUB characteristics. The correlation analyses of nucleotide contents did not reveal the role of natural selection by the host. These results suggest that mutational pressure due to compositional constraints shapes the SCUB in MPXV genomes to a large extent.
Spearman rank correlation analysis between overall and silent base compositions.

P < .05.
P < .01.
Quantification of SCUB
The ENC versus GC3 plots were developed to quantify the SCUB (Figure 1). The ENC values were found to vary by 47.00 ± 0.078. The calculated ENC values of all genes were found to be greater than 35, suggesting a weak codon bias in all examined MPXV genomes. The ENC values were approximately normally distributed, and the Mann-Whitney 2-sample test revealed no significant intergenomic differences in the ENC values (P > .05). In the plots, most of genes were found to lie on or just below the expected GC3 curve, suggesting that the SCUB was shaped mainly by GC compositional constraints. However, a considerable number of genes were grouped far below the expected GC3 curve, suggesting that other factors also influenced the SCUB in the MPXV genomes.
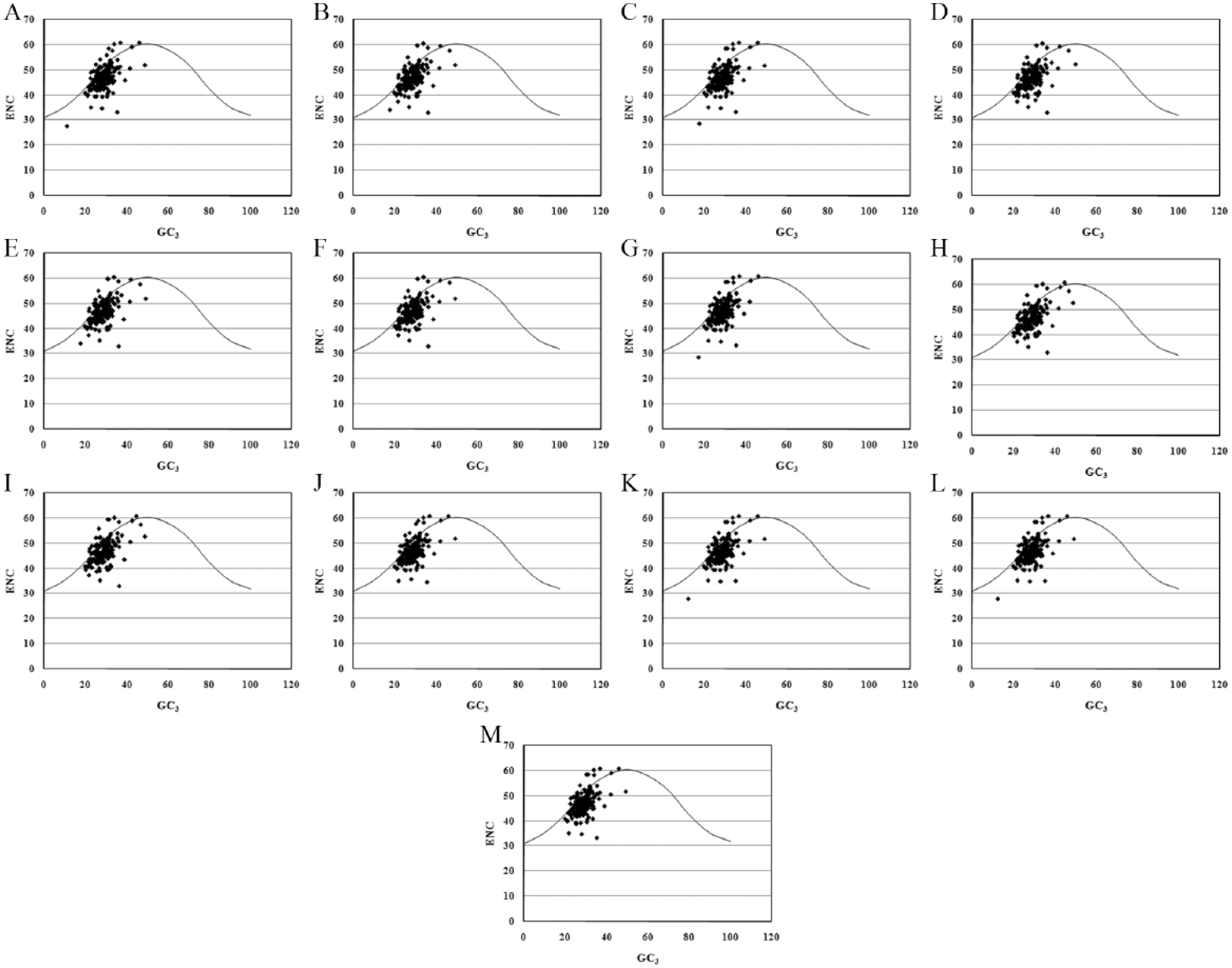
Mutational pressure versus selection pressure in MPXV genomes. ENC versus GC3 plots for (A) Congo-2003-358, (B) COP-58, (C) DRC Yandongi-1985, (D) Liberia-1970-184, (E) MPXV-WRAIR7-61, (F) Sierra Leone, (G) Sudan-2005-01, (H) USA-2003-039, (I) USA-2003-044, (J)V79-I-005, (K) Zaire-1979-005 (cr), (L) Zaire-1979-005, and (M) Zaire-96-I-16. ENC indicates effective number of codons; MPXV, monkeypox viruses.
Neutrality plots 65 revealed no significant correlations between GC3 and GC12 (the G and the C contents at the first and the second codon positions) as the slope of the scatterplot approached 0, which is an indication that other major factors, such as selection, also have an influence on the SCUB in the MPXV genomes (Figure 2). The association between purines (A and G) and pyrimidines (C and T) was analyzed using a PR2 bias plot, and the A and the T contents were found to be used more than the C and the G contents (Figure 3). The PR2 bias plots clearly exhibited deviations from Chargaff’s second parity rule 66 as most of the genes were localized far from the origin of the axis (Figure 3). The values of the PCG in all analyzed MPXV genomes (Table 1) had CAI values greater than 0.50; this indicated good host adaptation as the CAI values were calculated based on the Homo sapiens general codon usage. Significant positive correlations were found between the ENC and the CAI (P < .05), indicating that the level of gene expression had a large influence on the SCUB. The ENC was also positively correlated with the GC3 values (P < .01) and with the hydrophobicity scores (P < .05), revealing their crucial roles in shaping the SCUB in MPXV genomes.
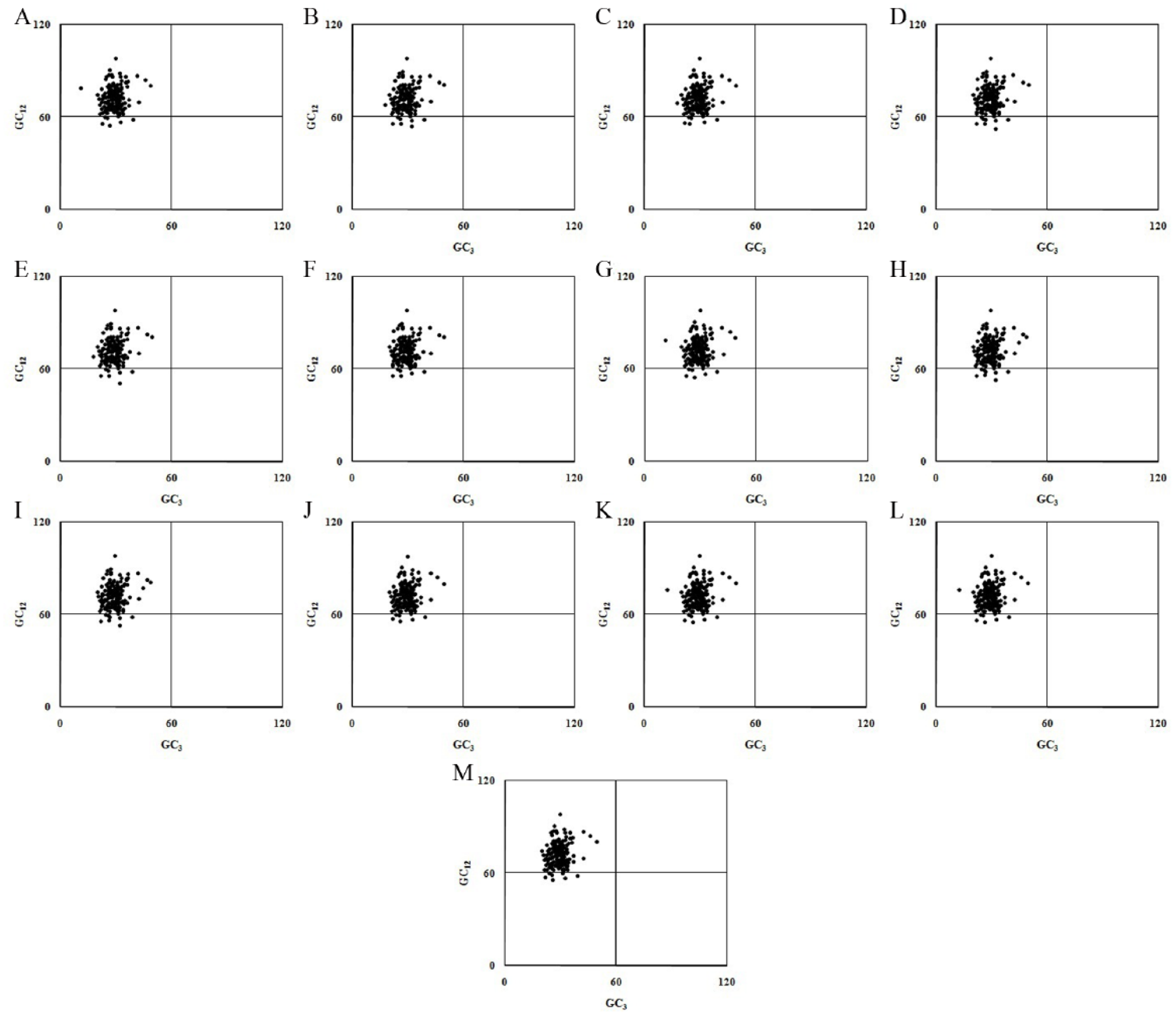
Influence of GC in shaping SCUB in MPXV genomes. Neutrality plots for (A) Congo-2003-358, (B) COP-58, (C) DRC Yandongi-1985, (D) Liberia-1970-184, (E) MPXV-WRAIR7-61, (F) Sierra Leone, (G) Sudan-2005-01, (H) USA-2003-039, (I) USA-2003-044, (J)V79-I-005, (K) Zaire-1979-005 (cr), (L) Zaire-1979-005, and (M) Zaire-96-I-16. MPXV indicates monkeypox viruses; SCUB, synonymous codon usage bias.

Deviation from parity rule 2 in MPXV genomes. PR2 bias plots for (A) Congo-2003-358, (B) COP-58, (C) DRC Yandongi-1985, (D) Liberia-1970-184, (E) MPXV-WRAIR7-61, (F) Sierra Leone, (G) Sudan-2005-01, (H) USA-2003-039, (I) USA-2003-044, (J)V79-I-005, (K) Zaire-1979-005 (cr), (L) Zaire-1979-005, and (M) Zaire-96-I-16. MPXV indicates monkeypox viruses.
Qualitative evaluation of SCUB
The codons with RSCU values greater than 1.0 are considered to be preferred as such codons are used more often than those with RSCU values less than 1.0. 3 In all synonymous amino acid families (6-fold, 4-fold, 3-fold, and 2-fold degenerate amino acids), A/T-ending codons were found to be used more frequently than G/C-ending codons (Table 3). In contrast, the human cells (host) use G/C-ending codons more frequently than A/T-ending codons.67,68 The AGA that codes Arg is the only A-ending codon preferred in human cells.67,68 In MPXV genomes, GAC (D), GGG (G), GGC (G), CAC (H), ATC (I), AAG (K), CTC (L), CTG (L), AAC (N), CAG (Q), AGG (R), CGC (R), AGC (S), TCC (S), ACC (T), ACG (T), GTG (V), GTC (V), and TAC (Y) were noted to be rare (RSCU < 0.66). In the host genome, the rare codons were reported to be GCG, CGA, AAT, GAT, TGT, CAA, GAA, GGT, CAT, ATA, TTA, AAA, TTT, CCG, TCG, ACG, TAT, and GTA.67,68 Strand-specific codon biases were observed in all MPXV genomes for the amino acid Ile; ie, in positive strands, all strains preferred ATA, whereas in negative strands, all strains preferred ATT (Table 4). The amino acids Arg, Thr, and Val also exhibited strand-specific bias, but not in all strains (Table 4). Interestingly, positive strand–encoded genes preferentially used A-ending codons, whereas negative strand–encoded genes preferred T-ending codons. However, in the negative strand–encoded genes of the DRC Yandongi-1985 and the Sudan-2005-01 strains, the amino acid Val preferred both GTT and GTA.
Overall relative synonymous codon usage values of protein-coding genes in examined monkeypox virus.

Abbreviations: AA, amino acid; MPXV, monkeypox viruses.
Codons exhibiting strand-specific bias in examined monkeypox virus genomes.

Abbreviations: AA, amino acid; MPXV, monkeypox viruses.
Bias in the dinucleotide frequency analysis demonstrated that AT was overrepresented in all genomes, whereas GC was underrepresented. The ρ values of the dinucleotides were calculated by taking the ratio of the observed to the expected dinucleotide frequency and, in all genomes except GC, were found to be very close to 1. The most biased dinucleotides were ρAT, ρGA, and ρTC. The χ2 test revealed that the dinucleotide frequencies were not randomly distributed (P < .05).
Putative optimal codons were chosen based on the χ2 analysis of the 2 data sets formed by selecting 10% of the genes located at the 2 extremes of COA axis 1. All putative optimal codons were found to end in A/T (Table 5). The SCUBs of strains having threshold fitness or “good fitness” 24 were hypothesized to be shaped due to natural selection by the host. 24 However, the presence of A/T-ending putative optimal codons in the MPXV genomes, as found in this study, can be explained largely by the high AT content in the respective genomes. Natural selection by the host, if it existed, would have resulted in particular codon usage patterns in which amino acids would have preferentially used any nucleotide-ending codons. 24
Identified putative optimal codons in examined monkeypox virus genomes.

Abbreviation: MPXV, monkeypox viruses.
Various factors influencing SCUB
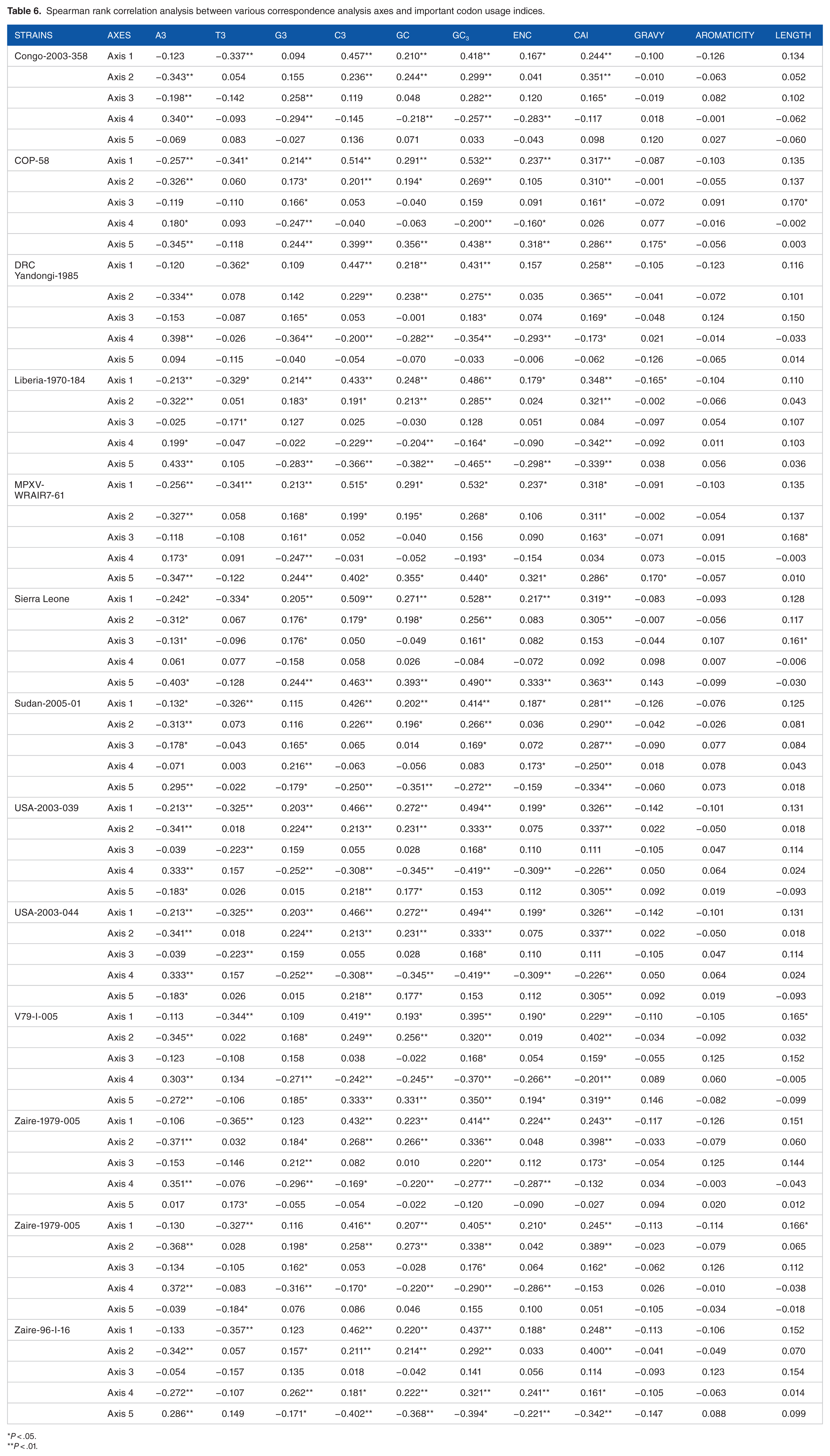
The COA partitioned the total number of SCU variations into 59 axes. Among the 59 axes, axes 1 to 5 accounted for approximately 10.42%, 8.43%, 7.13%, 5.66%, and 4.55% of the total SCU variations, respectively (Supplementary Figure 1). In all the strains isolated from various regions of Central Africa, E3 and GC3 had a high positive correlation with axis 1 (P < .01). The index indicating the level of gene expression (ie, CAI) had a higher positive correlation with axis 1 (P < .01) in all strains than the other proposed gene expression index, ENC, did (Table 6). The lengths of the coding sequences were weakly correlated with axis 1 for Central African strains such as V79-I-005 and Zaire-1979-005 (cr) (P < .05). The T3 content exhibited a significant negative correlation with axis 1 (P < .01) in all Central African strains (Table 6).
Spearman rank correlation analysis between various correspondence analysis axes and important codon usage indices.
P < .05.
P < .01.
In strains isolated from West Africa, the A3 content was highly negatively correlated with axis 1, whereas it was not correlated with axis 1 in strains from Central Africa. High positive correlation was observed between axis 1 and the G3 content (P < .01) for all West African strains. Similarly, G3 positively correlated with axis 1 in strains isolated from the United States. However, no correlation between G3 and axis 1 was observed in Central African and North African strains (Table 6). The CAI and the ENC also correlated highly with axis 1 in strains from West Africa and the United States (Table 6). That GC3 was significantly correlated with the first principal axis (ie, axis 1 in all strains) strongly suggests that nucleotide compositional constraints play an important role in shaping the SCUB across all MPXV genomes. Furthermore, high positive correlation with CAI (P < .01) revealed that the level of gene expression might also influence the SCUB across the examined MPXV genomes.
A correlation analysis between the dinucleotide content and the various COA axes did not reveal any true SCUB features, although some correlations did exist (Table 7). A cluster analysis of the pooled RSCU values of the PCG for each strain revealed 2 major clusters (Figure 4). More virulent Central African strains formed the upper cluster, and less virulent West African strains formed the lower cluster, indicating the presence of SCUB variations based on epidemic region and virulence.
Spearman correlation analysis between various correspondence analysis axes and dinucleotide contents.

P < .05.
P < .01.

Relationship between synonymous codon usage bias and virulence. The cluster analysis grouped more virulent strains into one major cluster (upper cluster) and less virulent strains into another cluster (lower cluster). CAI indicates codon adaptation index; ENC, effective number of codons.
Discussion
In this study, trends associated with the SCUB and with various factors influencing its diversification in selected MPXV genomes were investigated in detail. Studies related to the evolution of MPXV genomes are highly important as MPXVs can be used as potential bioterrorism agents. 69 The mean ENC values of all examined MPXV genomes were greater than 40, indicating weak SCUB. The weak MPXV bias may be attributed to the ability of an MPXV to suppress antiviral CD4+ and CD8+ T-cell responses by inhibiting antiviral T-cell activation and inflammatory cytokine production without involving major histocompatibility complex molecules as this mechanism would reduce competition between the virus and the host, leading to efficient dissemination in the host. 70 Monkeypox virus infection effectively inhibits the genes involved in stimulating innate immunity, thereby suppressing the expressions of proteins such as TNF-α, IL-1α/β, CCL5, and IL-6. 71 Thus, these findings form the basis for the observed weak SCUB of the PCG across all examined MPXV genomes.
The SCUBs of all mammalian genomes are comparable, and all human viruses share this pattern of codon usage with the human host. 72 This sharing reveals the need for human viruses to adapt their codon usage to the host if the infection is to be successful, whereas in other mammalian viruses, adaptation is not a prerequisite for infecting the host. 72 Two possible scenarios, which form the basis for developing this phenomenon, are coevolution of humans and viruses infecting humans and/or evolution of a human genome from a viral genome. 73
Significant intragenomic variations in the ENC (SD > 4.0) and the GC3 (SD > 4.0) values were observed in all the MPXV genomes used in this research. This heterogeneity in the base composition suggests that base compositional constraints play an important role in shaping SCUBs in MPXV genomes. A similar heterogeneity in the base composition was reported in herpesviruses belonging to the family Poxviridae. 7 Strand-specific codon usage was observed in MPXV genomes, whereas in the host genome, tissue-specific codon usage was reported; that is, in humans, the SCUBs of brain-specific, liver-specific, uterus-specific, testis-specific, ovary-specific, and vulva-specific genes were different from one another. 74 The SCUB in an MPXV may not be due to the GC composition as no correlations were observed between the GC3 and the cumulative GC values at the first and the second codon positions. However, AT richness is directly linked with SCUB as most preferred codons were A/T ending. Gene length was weakly correlated with different COA axes in some MPXV genomes, for example, the West African genomes COP-58, MPXV-VRAIR7-61, and Sierra Leone with axis 3, and the Central African genomes V79-I-005 and Zaire-1979-005 with axis 1. In addition, based on our analysis using axis 1 of the COA (the principal axis explaining most of the variations), we suggest that gene length may have a significant influence on SCUB only in Central African strains such as V79-I-005 and Zaire-1979-005.
All putative optimal codons were found to be A/T ending as MPXV genomes are AT rich and GC poor. In MPXV genomes, genome-specific preference toward a certain subset of codons was observed. Four codons (GGA, GGT, TAT, and TTT) were used as optimal codons in most MPXV genomes, although some exceptions occurred. The overrepresentation of AT contents and the underrepresentation of GC contents in the MPXV genomes seem to be the reason behind the use of A/T-ending codons, rather than natural selection, being preferred by the host. The weak codon bias of most genes across all examined MPXV genomes suggests that selection for translational accuracy and speed has less influence in dictating SCUB, revealing an inability to act as expression vectors, as reported in herpesviruses, another class of large double-stranded DNA viruses. 7 However, the putative optimal codons identified in this study can be used for enhancing heterologous gene expression by increasing translational efficiency.7,75-78 Axis 1 of the COA and the CAI exhibited significant positive correlations in all examined MPXV genomes (P < .01), indicating that gene expression levels have profound influences on SCUB.
Although no dinucleotide contents were found to be in high correlation with axis 1 of the COA in any of the examined MPXV genomes, AT dinucleotides were overrepresented, whereas GC dinucleotides were underrepresented in all genomes; AT, GA, and TC dinucleotides were most biased as their ρ values were greater than 1.10. Because GC dinucleotides possess the highest thermodynamic stacking energy,23,79,80 viral genomes are always under selection pressure to decrease the GC dinucleotide frequency20,79,81 to enhance viral genome replication and transcription. 79 Unmethylated GC in viral genomes stimulates immune responses in the host. 82 Hence, to reduce antiviral responses from the host, viral genomes contain fewer GC dinucleotides. 20 The Spearman rank correlation analysis revealed high positive correlations between C3 and GC3 and the principal axis (axis 1) of the COA and a significant negative correlation between T3 and axis 1. These correlations suggest that base compositional constraints play a crucial role in dictating SCUB. Axis 1 was not correlated with aromaticity in any MPXV, indicating that aromatic amino acids do not have a special role in framing SCUB, which further reveals that all amino acids contribute to SCUB.
Protein hydrophobicity scores were weakly correlated with axis 1 in Liberia-1970-184. Moreover, Central African and West African MPXV genomes are genetically distinct. 47 Cluster analysis showed clustering of Central African strains and one North African MPXV strain (Sudan-2005-01) into an upper cluster with similar SCUBs, whereas other strains isolated from West Africa and the United States formed a lower cluster with similar SCUBs. However, the lower cluster revealed that the US-isolated MPXVs possessed similar SCUBs as they are in one clade close to Liberia-1970-184. Furthermore, Central African strains have been reported to be more virulent than West African strains. 47 Based on these results, we are able to postulate that a strong association exists between MPXV strain virulence and SCUB as more virulent strains formed one cluster exhibiting similar SCUBs, and less virulent strains formed another. Thus, we conclude that mutational pressure due to base compositional constraints, level of gene expression, and codon selection for utilization of putative optimal codons are major factors influencing the SCUB in MPXV genomes. Consequently, a balance exists between mutational pressure acting on nucleotide sequences and amino acid selection in MPXV genomes, which is similar to the finding in a report on hepatitis E viruses. 1 Generally, to conserve the protein sequence, purifying selection eliminates transversions at the third codon positions in 2-fold degenerate amino acids. Among the 20 amino acids, most synonymous positions are in 2-fold degenerate amino acids. Hence, selection may act on an amino acid level to eliminate the possibility of nonsynonymous transversions in 2-fold degenerate amino acids. In addition, viral genomes have naturally evolved with a mechanism to tackle and escape host antiviral responses, 28 and according to the evolution rhetoric theory, 83 this mechanism may also act as a major selection pressure in framing the SCUB in MPXV genomes, as reported in hepatitis A viral genomes. 28 In this context, the multifactorial codon usage bias in MPXV genomes might have evolved as the result of a need to increase the efficiency of communication from the genome to the cell in transitional environments by keeping the message unmodified.28,83
Supplemental Material
EVB761368_Supplementary_Material_REV1 – Supplemental material for Evolution of Synonymous Codon Usage Bias in West African and Central African Strains of Monkeypox Virus
Supplemental material, EVB761368_Supplementary_Material_REV1 for Evolution of Synonymous Codon Usage Bias in West African and Central African Strains of Monkeypox Virus by Sudeesh Karumathil, Nimal T Raveendran, Doss Ganesh, Sampath Kumar NS, Rahul R Nair and Vijaya R Dirisala in Evolutionary Bioinformatics
Footnotes
Acknowledgements
Language editing of this manuscript was provided by Edward J Button, PhD, CEO, Button and Associates, VA, USA. The first author (S.K.) would like to thank Dr TP Jayakrishnan (Director of Aushmath Biosciences) for providing support for the successful completion of this study.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
RRN conceived the idea and designed the methodology. SK, NTR, and GD performed the analyses. SK, RRN, VRD, GD and SKNS interpreted the results. RRN wrote the manuscript. GD, SKNS and VRD offered critical comments. RRN and VRD developed the final draft. All authors read and approved the final manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.