
Abstract
Three-dimensional models of the alpha- and beta-1 subunits of the calcium-activated potassium channel (BK) were predicted by threading modeling. A recursive approach comprising of sequence alignment and model building based on three templates was used to build these models, with the refinement of non-conserved regions carried out using threading techniques. The complex formed by the subunits was studied by means of docking techniques, using 3D models of the two subunits, and an approach based on rigid-body structures. Structural effects of the complex were analyzed with respect to hydrogen-bond interactions and binding-energy calculations. Potential interaction sites of the complex were determined by referencing a study of the difference accessible surface area (DASA) of the protein subunits in the complex.
Introduction
Large-conductance, Ca2+-activated potassium (BK) channels physiologically fulfill various functions, such as the regulation of action potential duration and firing frequency in many neurons, 1 endocrine secretion,2,3 vascular smooth muscle tone, 4 tracheal smooth muscle, 5 and microbicidal processes. 6 The BK channel is a complex made up of four alpha and four beta subunits. 7 The alpha subunit, considered to be the pore-forming unit, consists of a core region, which includes hydrophobic segments S0–S6, and a long carboxyl extension containing the hydrophobic segments S7–S10. 8 The beta subunit is a regulatory subunit that includes two membrane-spanning segments, with the NH2 and COOH termini located on the intracellular side of the channel. So far, four different beta subunit subtypes have been identified (beta1, beta2, beta3, and beta4) 9 and they are differentially expressed in different tissues. 10 Thus, each beta subunit has a specific tissue distribution and different effects on BK channel pharmacology and activation gating. 10 Beta1, the most well studied subunit, is expressed in smooth muscle and plays an essential role in the normal regulation of vascular smooth muscle contractility and in blood pressure.
The absence of the beta1 subunit, inadequate coexpression, or the malformation of heteromultimeric BK channels from alpha and beta1 can modify the resulting channel's proposed role as negative feedback regulator of vascular tone. 11 Malfunctioning BK channels can lead to a wide variety of disorders, including epilepsy, motor impairment, noise-induced hearing loss, hypertension, urinary incontinence, overactive urinary bladder, and asthma [reviewed in 12 ]. While much is known about the structure, composition, and function of the BK channel, the nature of the physical link between the alpha and beta1 subunits is poorly understood. Based on studies of chimeras between the beta1 and beta2-IR subunits, Orio and colleagues 13 found that the NH2 and COOH termini of the beta subunits are relevant regions in the functional coupling between alpha and beta subunits. Other studies have demonstrated that the physical association between alpha and beta1 requires the S1, S2, and S3 transmembrane helices, but not the NH2 terminus of the alpha subunit. 14 It was also suggested that the extracellular ends of beta1, TM2, and S0, are in contact and that beta1 TM1 is close to both S1 and S2 in the alpha subunit. 15 Nevertheless, there is much about the structure of the BK channel that remains to be elucidated, such as identification of the residues involved in the physical association between its alpha and beta subunits.
Protein–protein interactions are often not accessible to experimental study because of their low stability and the difficulties involved in producing the proteins and to assemble them in their native conformations. Thus, docking algorithms have been developed to provide an in silico approach to the problem. The aims of this study were: (1) to predict the binding sites that link the alpha and beta1 subunits, using computational modeling and molecular docking; (2) to evaluate helix-helix interactions in pairs of helices; and (3) to generate most-probable models for the alpha and beta1 subunits, in both the monomeric and the assembled form. The results showed the highest probability of interaction for amino acids E147, E149, Y263, Y113, N201, and N237 of the alpha subunit, and amino acidsK3, K4, Y31, Y32, Q62, Q91, and Q113 for the beta1 subunit.
Materials and Methods
Definition of the TM helices
Nine secondary-structure prediction programs were used to predict the length of each helical stretch of the subunits: DAS, ALOM2, PHDtm, TMAP, TMHMM2, TMPRED, TOPPRED2, SPLIT4, and HMMTOP2. Based on the results, a consensus prediction was calculated according to a simple majority vote type procedure. Alom2 and DAS are methods that focus on local properties of amino acid sequences to decide which sub-sequences are most likely to span the membrane, usually in sliding Window approach. PHD uses a neuronal network and should still be regarded as a local approach. Global approaches are HMMTOP and TMHMM, which both implement circular Hidden Markov Models. These approaches determine the statistically most probable topology for the whole protein according to the underlying model. TMAP, Toppred2 and SPLIT4 represent combined forms, in which results on a local level are evaluated by global heuristics such as the positive-inside rule or other differences in the distribution of amino acids.
Molecular Modeling of the BK Channel's Alpha and Beta Subunits
The sequence of the human BK potassium channel was obtained from the Swiss-Prot database at http://expasy.org/sprot. 25 BLAST (basic local alignment search tool) 26 sequence analysis was performed against the whole protein data bank to detect homologous/analogous protein templates by matching the whole-chain sequences of target subunits to solved protein structures. Since conserved-structure pieces excised from the different protein structures were directly used to assemble the new protein-structure models, the threading modeling approach 28 was chosen together with the HHpred program.29,30 The prediction was made by constructing a structure model by placing the backbone atoms of the target sequence at their aligned backbone positions of the selected structural templates (PDB IDs: 3naf, 1k4c and 1f6g).
HHpred allows the user to test several alternative alignments and to evaluate the quality of the resulting models in order to achieve an optimal result. Results files contain the superposed template structures, and the alignment between each subunit file and template file are generated inside the HHpred program. 31 After following this approach, the generated alpha and beta1 subunits were validated using the PROCHECK program 32 and compared with the previous secondary structure predictions for the helix of each subunit at the level of the length of the secondary structure. The aim of PROCHECK is to assess how normal or, conversely, how unusual the geometry of the residues in a given protein structure is when compared to the stereochemical parameters of well-refined and high-resolution structures.
Protein–Protein Docking
The alpha and beta subunit structures derived from the secondary and tertiary structure predictions were each used as the starting structure in docking simulations with ClusPro's 33 as well as in GRAMM 34 algorithms for obtaining the bound structures of the alpha and beta1 subunits. ClusPro has the option of selecting either DOT35,36 or ZDOCK 37 to perform rigid-body docking and both are based on fast Fourier transform (FFT) correlation techniques.38,39 DOT was selected for the present work, since it allows for the use of an electrostatic potential in the scoring function and in the surface complementarities between the two structures. DOT runs on a 128A × 128A × 128A grid, with a grid spacing of 1Å. It performs 13,000 rotations, initially obtaining over 2.7 × 10 10 structures and finally retaining only 20,000 structures with the best surface complementary scores. These docked structures are then filtered using distance-dependent electrostatics 40 and an empirical potential energy. 41 The 2000 conformations retained after filtering are clustered based on the pairwise RMSD (root mean square deviation) 42 and the best conformational structure is selected. Finally, the representative conformation selected is refined using CHARMM minimization. 43 The GRAMM program was used because it runs in helix mode, ie, the search can be limited to helix pairs in antiparallel and parallel orientations, discarding configurations with large displacements along the helix axes and crossing angles larger than 10°.
The best structural model for the complex of alpha and beta subunits obtained from the docking simulations was subjected to MD simulation to refine the protein interface. However, no explicit constraint functions were used to maintain the initial docking contacts during the simulation. The structures were first energy minimized using 1000 steps of steepest descent and 2000 steps of conjugate gradient minimization implemented in GROMACS. A distance dependent dielectric function was used with the dielectric constant set to 1 and the non-bonded cutoff was set to 8Å. Energy minimization with classical force field can be used to remove unrealistically close steric clashes and large deviations from ideal geometry resulting from the conformational changes of amino acid side chains after docking. The binding free energy was calculated by MM-PBSA implemented in Amber 7.0.
Clustering on the Basis of Pairwise RMSD
Contact residues within 10Å of any atom of the fixed subunits at the interface conformations were analyzed. The measure employed here was not affected by those parts of the molecule far from the interface.
Computation of Individual Residue–Residue
Binding affinity, desolvation free energy, and electrostatic and contact free energies of docked structures were calculated using the program FastContact, 44 which provides an estimate of the binding affinity between two proteins. Helix interaction tool algorithm was also used for analysis of helix-helix interactions. 52
Results and Discussion
Given the lack of any direct experimental evidence related to the BK channel's secondary or tertiary structure, our task set was similar to the protein-folding problem. 45 Accordingly, we proceeded in a hierarchical manner. First, a helical character of the transmembrane parts of the protein was assumed based on the experimentally derived structure of the BK channel (PDBID: 1NAF). 35 The X-ray crystal structure of Calcium-activated potassium channel subunit alpha-1 was solved at 3.1Å resolution and found to be 798 amino acids in length. This structure reveals four intracellular subunits, each comprising of two tandem RCK domains, assembled into a gating ring. Second, multiple secondary prediction methods were used to determine the extent of each helix. Third, and finally, the structures of the alpha and beta subunit helices were predicted by generating a number of models, using either a threading protocol or a helix-helix docking approach.
Nine secondary structure prediction programs were used to predict the length of each helical stretch of the alpha and beta subunits (Fig. 1). The length of the helix of the alpha subunit (Fig. 1A) varied considerably depending on the prediction method. DAS predicted a helix with a size ranging from 1 to 22 amino acids. The other programs, TMAP, TMHMM2, TMPRED, suggested helices with lengths of 18–25 amino acids; SPLIT4 helices were 20–30 amino acids long; and TOPPRED2, PHDtm, ALOM2, and HMMTOP2 helices consisted of 20, 17, 16, and 18 amino acids, respectively.

Consensus of secondary structure predictions for the alpha and beta1 subunits. (
For the beta1 subunit, the shortest sequence was predicted by DAS, with 23 amino acids, followed by ALOM2, which predicted a 16-residue helix. The helices predicted by the other programs were 18–22 amino acids in length. TMAP was the only program that did not discriminate between the two helical regions and instead suggested a long helical stretch of 26–28 amino acids. From the above results, a consensus prediction was calculated according to a simple majority vote type procedure (Fig. 1B). If more than seven methods predicted a residue to be helical, it was assigned as such.
Based on the secondary-structure prediction, the tertiary structures of the alpha and beta subunits were modeled by computational methods. We used molecular docking and helix interaction tool for analysis of helix-helix interactions. 52 While the two docking methods used cannot consider the structural flexibility of the binding site during the docking process, the most favorable solution obtained was then refined through molecular dynamics to get the final docked model (Fig. 2).
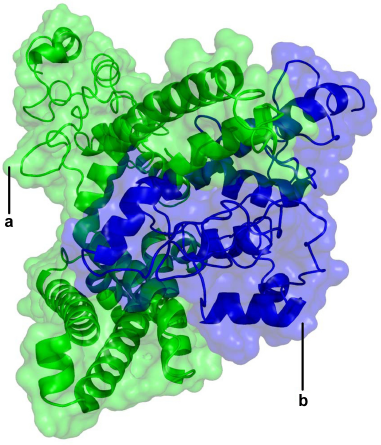
Three-dimensional structure image according to the molecular docking simulations.
The selection of the final model for the docked complex was based on factors such as the area of surface contact, extent of interactions, and difference in the binding free energies for complex conformations. In the MM-PBA calculation of the binding free energy for the most favorable solution obtained was calculated to be –13.7 Kcal/mol.
It was found that residues K3, K4, and R11 in the NH2-terminal domain of the beta1 subunit interact with residues E147 and E149 in the S0–S1 intracellular loop of the alpha subunit (Fig. 3). Additional interactions were between amino acids K179, Y183 in the COOH terminus of the beta1 subunit and amino acid N237 from the S2–S3 intracellular loop of the alpha subunit (Fig. 4). These residues may provide electrostatic interactions and involve hydrogen bonds between NH2 and COOH segments of the regions. It is worth noting that these results are close to those previously reported by Yang et al, 14 who noted the importance of residues N237 (N172 in Yang et al) 14 and D165 (D99 in 14 ) in BK channel activation. Although our results did not show a direct interaction with D165, they did predict interactions with the nearby residues E147 and E149, located in the S0–S1 linker. Yang and colleagues 14 proposed a model in which these residues form a binding site for Mg2+, generating an electrostatic interaction with residue R288 (R213) to activate the voltage sensor. These results suggest that interactions between the NH2-and COOH-terminal segments of the beta1 and alpha subunits help to position these intracellular domains near the RCK1 domain, which is directly involved in voltage sensor activation. Recently, using chimerical proteins between dslo1 and mslo1, Lee et al 46 proposed that linker S0–S1 is an important region potentially involved in the increase of calcium sensitivity that beta 2 induce on alpha subunit. Related with beta subunits, Orio et al 13 pointed out the importance of the NH2-terminal domain of this subunit with respect to the modulation of voltage and calcium sensitivity on BK channel. Similarly, Wand and Brenner 47 reported that deletion of both the amino and the carboxy segment of the beta1 subunit removed the shift in the G-V relation induced by this subunit. These results support the importance of these domains in alpha and beta subunit interactions, probably by a direct interaction between these domains and the region responsible for BK channel activation. Extending these findings, it would be of interest to create mutations in residues K3, K4, R11, K179, and Y183 of the beta1 subunit and in residues E147, E149, and N237 of the alpha subunit.

Interaction between the NH2-terminal intracellular β1 and the S0–S1 intracellular loop; and between the S3–S4 extracellular loop of the α subunit and TM2 of the β1 subunit. The α and β1 subunits are shown in green and blue, respectively. (
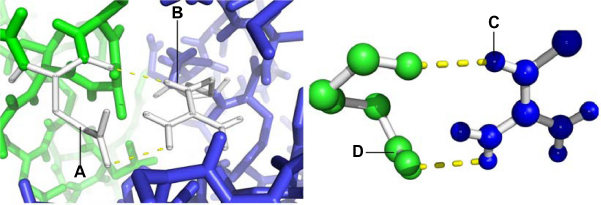
NH2-terminal intracellular β1 interacting with the S0–S1 intracellular loop; and between the S3–S4 extracellular loop of the α subunit and TM2 of the β1 subunit. The α and β1 subunits are shown in green and blue, respectively. (A) Glutamic acid 149 (B) Arginine 11.
Other potential sites of interaction were identified between different residues of the extracellular loop of the beta1 subunit (Q63, Y74, Q99 and D111) and an amino acid located in the S1-S2 extracellular linker of the alpha subunit (N201) (Fig. 5 and Table 1). Previously, Hanner et al 15 described the relevance of several residues in the extracellular loop of the beta1 subunit with respect to interactions with the alpha subunit. Contrary to our predictions, they reported that residues Q99 and D111 do not play a direct role in the interaction between the two subunits, suggesting that despite the high probability of interaction between these residues and amino acid N201, additional interactions are necessary in other regions of the beta1 subunit to generate a proper link between it and the alpha subunit. An alignment analysis of the sequences of the beta subunit subtypes (beta1, beta2, beta3, beta4) showed that residue Q63 is not conserved, unlike residue Y74, which is present in all BK beta subunits. This is consistent with the important role postulated for this residue in the coupling between the alpha and beta1 subunits of BK channels and involving a hydrogen bonding interaction with residue N201 of the alpha subunit. Thus, residue Y74 of alpha subunit would also be an interesting target for mutation.

Interactions NH2 and COOH terminal of the β1 subunit.
Interacting residues in the alpha and beta subunits

Another coupling site was found between residues located in the S1 transmembranal domain (S199, Y195) of the alpha subunit and the TM1 domain (Y31) of beta1 (Fig. 5). This interaction is consistent with the findings of Liu et al, 48 who mutated the first four residues from extracellular loops of both alpha and beta1 by changing each one to a cysteine and thus found a close relationship between S1 and TM1. The interactions established between residues (S199, Y195 to Y31) are predicted to be in the form of hydrogen bonds between hydroxyl-hydroxyl groups, which are very stable when located in the channel's hydrophobic interior.
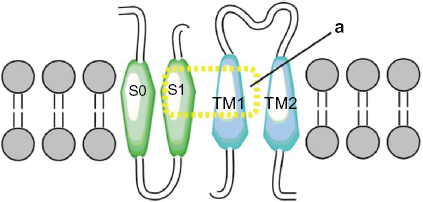
Two more interactions were identified. The first was between the S3 and S4 extracellular loop (Y263) of the alpha subunit and the extracellular loop (N142) of the TM2 domain of the beta1 subunit. The second one predicted an interaction between the S3 and TM2 domains (Fig. 6). These results differ from those of Liu and colleagues7,39 who reported that the extracellular ends of beta1 TM2, and S0 are in contact. However, our findings agree with the results of Morrow et al, 40 who reported that the beta1 subunit interacts with the S3 transmembrane (Fig. 7) domain exclusively; without excluding the possibility that the topology of the S1 and S2 domains is affected by expression of the S3 domain (Fig. 8). Therefore, we propose a theoretical model in which TM2 is close to S3, and the S0 domain is located between S2 and S3 (Fig. 9).

Interaction loop extracellular β1.
Interaction transmembrane TM1 and S1.

Interaction TM2 of the α subunit. (

Theoretical model in the figure represent two α subunits (green), two β1 subunits (blue), theoretical model TM2 is close to S3 and S0 domain is between S2 and S3.
Conclusions
The results of this study suggest a physical-link model in which specific residues interact to induce the coupling of the alpha and beta1 subunits of BK channels. Previous reports using chimerical proteins and deletions have shown that NH2 and COOH termini domains in the beta subunit, as well as the intracellular loop S0–S1 in the alpha subunit, are important for functional properties like voltage and calcium sensitivity of the channel. Our computational approach was in agreement, indicating that specific residues located in NH2 (K3, K4 y R11) and COOH (K179, Y183) termini of BK beta subunit and some residues on intracellular loop S0–S1 in BK alpha subunit (E147, E149) are important for physical interaction between both subunits. Taking this into account, we propose the residues K3, K4, R11, K179 and Y183 in beta1 subunit as well as E147 and D149 in alpha subunits are possible candidates for point mutations that allow us to analyze the true importance of the respective residues and their contribution to the physical interaction between these subunits.
Funding Sources
This work was funded by Pontificia Universidad Javeriana, Project: “Estudio de la interacción del dominio amino terminal de la subunidad beta1 con la subunidad alfa del canal de potasio de alta conductancia dependiente de Ca2+ y voltaje (BK)” (ID 3262).
Author Contributions
Conceived and designed the experiments: YT, JG. Analyzed the data: AG, OS. Wrote the first draft of the manuscript: JG. Contributed to the writing of the manuscript: GB. Agree with manuscript results and conclusions: LM. Jointly developed the structure and arguments for the paper: JG, GB. Made critical revisions and approved final version: FC. All authors reviewed and approved of the final manuscript.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Footnotes
Acknowledgements
The authors thank the Pontificia Universidad Javeriana for providing the necessary facilities to carry out this work.