
Abstract
In this study, we investigated the modalities of coding open reading frame (cORF) classification of expressed sequence tags (EST) by using the universal feature method (UFM). The UFM algorithm is based on the scoring of purine bias (Rrr) and stop codon frequencies. UFM classifies ORFs as coding or non-coding through a score based on 5 factors: (i) stop codon frequency; (ii) the product of the probabilities of purines occurring in the three positions of nucleotide triplets; (iii) the product of the probabilities of Cytosine (C), Guanine (G), and Adenine (A) occurring in the 1st, 2nd, and 3rd positions of triplets, respectively; (iv) the probabilities of a G occurring in the 1st and 2nd positions of triplets; and (v) the probabilities of a T occurring in the 1st and an A in the 2nd position of triplets. Because UFM is based on primary determinants of coding sequences that are conserved throughout the biosphere, it is suitable for cORF classification of any sequence in eukaryote transcriptomes without prior knowledge. Considering the protein sequences of the Protein Data Bank (RCSB PDB or more simply PDB) as a reference, we found that UFM classifies cORFs of ≥200 bp (if the coding strand is known) and cORFs of ≥300 bp (if the coding strand is unknown), and releases them in their coding strand and coding frame, which allows their automatic translation into protein sequences with a success rate equal to or higher than 95%. We first established the statistical parameters of UFM using ESTs from
Introduction
With the arrival of third generation methods for DNA sequencing, we are facing a new reality in which high-throughput sequencing is becoming affordable. This means that high-throughput data mining is necessary now more than ever. 1 With this concern in mind, the transcriptome is the first component to be addressed when approaching a new large genome, for example, that of a plant. 2 Because the transcriptome is the expressed part of a genome, it is a representation of the genome's coding potentialities. One must first consider that codon usage is specific to the species under investigation and is the feature that is generally considered by gene classifiers. Secondly, in prospective research, the information on codon usage for model training is not necessarily available or transferable from one species to the other. Thus, a method based on features that would allow coding open reading frame (ORF) classification independently of the codon usage is desired. This process is the same as the automatic extraction (or classification) of coding ORFs (cORF) from large samples of expressed sequence tags (EST) or full complementary DNA (cDNA) sequences. The question remains of how to automatically extract the coding component of the transcriptome to achieve high statistical significance and a low error rate, without the need for prior knowledge concerning the biological species under investigation.
Several methods for automatic extraction have previously been proposed:
23
OrfPredictor
4
(http://proteomics.ysu.edu/tools/OrfPredictor.html) provides six-frame translation and predicts the most probable coding regions among all frames; Diogenes, which is available from the University of Minnesota (http://www.ahc.umn.edu/), identifies ORF candidates by scanning all six frames for stretches of sequences uninterrupted by stop codons, codon frequency and ORF length, which are then used to estimate the likelihood that these ORF candidates encode proteins using a quadratic discriminant function combining these various factors; ESTScan
5
(http://www.ch.embnet.org/software/ESTScan.html) and DECODER,
6
which are based on hidden Markov models (HMM), can detect and extract coding regions from low-quality ESTs or partial cDNAs while correcting for frame-shift errors; DIANA-EST,
7
which is based on TIS prediction through neural network technology; and Prot4EST
8
(http://www.compsysbio.org/lab/?q=prot4EST), which is a pipeline that predicts and translates ESTs into polypeptides using DECODER, ESTScan, and the
Here, we chose the universal feature method (UFM) as an investigative tool. UFM combines statistics for stop codons, ancestral codons (RNY, where R stands for a purine, N for any of the four nucleotides and Y for a pyrimidine), and base usage over six frames in a hierarchical flowchart. 10 UFM's performance should not be compared with the performance of other EST processing methods5-8 because none of these methods have a comparable cost benefit ratio. In addition, these alternative methods5-8 require prior training or parametric adjustment of a model for a species family, whereas UFM does not. Thus, choosing the largest ORF within a transcript is the only alternative to UFM that can be used as a control to measure comparative performance. UFM is the only method that combines the stop codon density and purine bias that are both criteria specific to coding DNA and independent of biological species. The performance of UFM in the classification of introns and coding sequences (CDS) has been shown to be both better than related methods available from independent investigations and to perform at the 95% level of success rate classification, over the whole spectrum of codon usage.10,11 Non-classified sequences (missing data) allow the evaluation of UFM classification in real situations and objective context, such as the transcriptome, which is the purpose of this study.
We found that the cORFs obtained via UFM classification of contigs from ESTs show a GC3 distribution similar to that of reference samples of CDSs. In addition, when translated into protein sequences and compared to PDB sequences using BLASTp, cORFs ≥ 300 bp produced 95% matches. These results suggest UFM can be used to extract the coding component of any transcriptome sequence set, without any previous knowledge of that species. Thus, according to the size limitation (cORFs ≥ 300 bp), UFM can be used in connection with CAP3 12 to classify cORFs from the transcriptome of any eukaryote species, without any previous knowledge of that species.
Methods
Sequence Materials and Experimental Scheme
Referencing highly confident samples of true positives becomes crucial when addressing the problem of classification optimization. To address this challenge without ambiguity, we used three strategies. First we gathered CDS samples with strong indication of being true positive, ie, ESTs homologous to the protein sequences of PDB (http://www.rcsb.org) since all accessions of PDB are: experimentally expressed, crystallized protein, and have a known function. Secondly, we gathered the EST samples among species that cover the whole range of codon usage, ie, the whole GC3 (the guanine plus cytosine content in 3rd position of codons) range of eukaryotes.10,11 Thus, ESTs from
Samples of the EST datasets just described (queries) were compared with protein sequences (subjects) from PDB (January, 2010) in homology searches using BLASTx. The file outputs were then parsed according to their respective informative fields (query name, query size, subject name, subject size, subject definition, score, bit score, expected, identity, similarity, gaps, first base of query homology, last base of query homology, first base of subject homology, last base of subject homology) using Perl. Entries with a
In this study we considered all nucleotide triplets between two stop codons as ORFs and all nucleotide triplets that could be drawn from the first 5′ in-frame ATG and the 3′ end of this ORF as
The sequences of EST regions homologous to PDB protein sequences were considered true positive CDSs, as they are expressed and contain a region that is homologous to a protein that has been crystallized (experimental data). These samples represented a convenient means of measuring the sensitivity and specificity of cORF diagnosis because the coding status and the exact position of the cORF in the sequences are known.
To find the size threshold at which a 95% success rate of cORF diagnosis occurred using the UFM classification, we set the minimum ORF size cutoff and the minimum size of the homologous regions between ESTs and protein sequences from PDB to the same value. This meant that when the EST sample contained sequences in which the region homologous to PDB was ≥100 bp, we implemented UFM with a cutoff of 100 bp; this is in contrast to when the EST sample contained sequences in which the region homologous to PDB was ≥200 bp, UFM only considered ORFs with a size ≥ 200 bp, and so on. Because the average size of ESTs homologous to PDB is rather small, samples with an average size larger than ~350 bp were not statistically consistent across all of the species addressed in this study. For this reason, UFM thresholds larger than 350 bp could not be considered for ESTs.
In order to compare the consistency of cORF sizes between species with different codon usages and species that belong to different phyla, we retrieved all CDSs of
When the statistical parameters of cORF classification among three and six frames by UFM were established, from the samples of ESTs via reference to the coordinates of PDB homologous regions, we tested the cORF classification produced by UFM in large samples of ESTs. For this purpose, we first extracted cORFs from the complete pools of ESTs in dbEST (GenBank, rel 175) for
The consistency of cORFs obtained from EST contigs was evaluated by comparing their GC3 distribution to those of reference samples of rice and human CDSs. In humans, the reference samples were those reported by Zoubak et al, 15 since a significant amount of work on human genome organization has been carried out based on that study. At the time, genes were mostly described via experimental procedures, and the known gene number was small. Therefore, the possibility of a bias for GC-rich sequences due to the small sample size of this dataset could exist, as GC-rich genes with few or small introns were easier to sequence because of their shorter size. 16 Today, most human genes have been described and their curated CDSs are available for comparative analysis. Thus, we also compared the GC3 distribution provided by Zoubak et al 15 to that of the CDS sample curated by Fedorov's group (n = 23,366), which is listed in the file hs37p1.EID.tar.gz and can be downloaded from http://www.utoledo.edu/med/depts/bioinfo/database.html.17,18 To link this CDS sample with experimental evidences, we compared the protein sequences of these CDSs with the protein sequences of PDB to determine their homology (E < 0.0001). The homologous hits were then filtered so that only the best hit was kept (n = 13,672) for each human accession. We then filtered the list, keeping pairs with an identity ≥ 40% (n = 10,892) and we used the accession identifiers to retrieve their corresponding DNA sequences from the original CDS file (n = 23,366). We considered this dataset (n = 13,672) as our sample of true positives.
The curated sample of human intron sequences (hs35p1.intrEID) that we used to measure the success rate of the CDS vs. non-CDS classification also came from Fedorov's group.17,18 For the purpose of comparison with CDSs, in this file we only considered the first 10,348 sequences larger than 300 bp. We considered this dataset (n = 10,348) as our sample of true negatives.
In the case of rice, the reference CDS distribution was that published by Carels et al, 19 which was obtained by certifying the coding status of the whole set of CDSs ≥ 600 bp from The Institute for Genomic Research (TIGR), according to the average mutual information (AMI). 20
Coding ORF Diagnosis
As explained above, putative cORFs were extracted from sequence samples using the UFM algorithm implemented as shown in Figure 1.
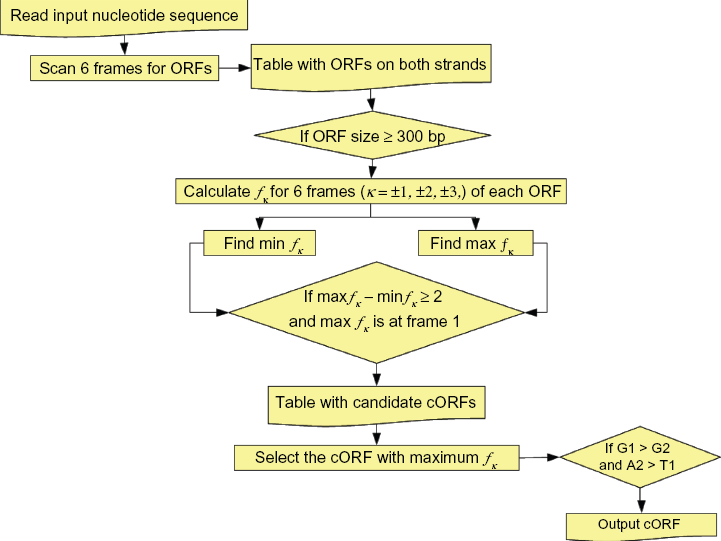
Flow chart of UFM applied to cORF finding in transcriptome sequences.
Error Evaluation
To evaluate the classification error associated with UFM, it is necessary to first consider that the error may concern 3 factors—the diagnosis of the coding status (coding or not coding), the coding strand, and the coding frame. These three components each have specific false positive, true positive, false negative, and true negative rates. Here, we explain how we measured their rate by reference to the regions of ESTs homologous to PDB protein sequences, which are considered as true positive sequences, strands, and coding frames.
The Coding Frame
If a cORF is on the same strand as the homologous region used as the true positive reference, there are 3 necessary conditions for that ORF to be considered as a true positive for the frame. First, the coordinate of the first base of the ORF must be smaller or equal to the coordinate of the first base of the homologous region because the homologous region is in frame with the coding sequence. Second, the coordinate of the last base of the ORF must be larger or equal to the coordinate of the last base of the homology detected by BLASTx. Third, there must be a whole number of nucleotide triplets (codons) between the first base of the ORF and the first base of the alignment because they must both be in frame with the coding sequence. Any ORF diagnosed as a cORF that does not obey these conditions is a false positive. False negatives correspond to the cases where a frame is diagnosed as false when it is true. This situation can only occur when a coding ORF is classified as non-coding; it is therefore a component of the false negatives with respect to coding status that are evaluated here via the success rate. The same situation occurs with true negatives.
The Coding Strand
The success rate of strand diagnosis is easy to evaluate by simply counting the number of times the strand of the homologous region matches that of the ORF (query). For example, “Plus/Plus” homologies are true positives, whereas “Plus/Minus” homologies are false positives. Again, true negatives and false negatives are part of the true negatives and false negatives regarding coding status, which was evaluated here based on the success rate of true positive detection.
The Coding Status
The rate of false negatives is given by the rate of cORFs not detected by UFM. The rate of true negative has been analyzed extensively elsewhere 10 and will not be revisited here.
Finally, we evaluated the performance of UFM by comparing it to the success rates of the classification of coding sequence, strand, and frame diagnosis of the largest ORF (LORF) of the sequence considered.
At this point, we can ask what would occur when a sequence does not share a BLASTx hit with PDB. Because of the statistical distribution, we can assume that the rate of true positives, false positives, true negatives, and false negatives will be the same in this case as in the model (ORFs showing homology to PDB).
Results
Highly confident samples of true positives are necessary to test and optimize algorithms for cORF classification. The search for homologies among ESTs with PDB sequences is a simple method for this purpose. The rate of in-frame stop codon occurrence in these homologous regions of ESTs was found to be <15% in
Search of homologies between ESTs from dbEST (GenBank) with the protein sequences of PDB using BLASTx (E < 10–4).

N is the sample size of EST sequences;
the numbers into brackets are percentages of sample size.
Coding Frame Classification when the Coding Strand is Known
Considering the success rate of coding frame detection, two situations can occur. Either the coding strand is known, for example, because the polyA tail has been detected, or the coding strand is unknown, because that information is not given.
In cases where the coding strand is known, the coding frame must be chosen from among three possibilities. The success rate of coding frame detection corresponding to such cases is given in Table 2. A 95% success rate of UFM in coding strand diagnosis (column “Bg&Phs” of Table 2) was achieved for ORF sizes ≥ 200 bp in higher eukaryotes (
Success rate of coding ORF diagnosis among the three frames of ESTs of

Sp: species name.
In comparison to diagnosis based on the largest ORF (LORF), diagnosis using UFM appeared to be more robust and stable across species. In lower eukaryotes, the sensitivity was clearly higher for LORF, but this is associated with a lack of discrimination because this method would also find a LORF in non-coding ESTs; thus the sensitivity is deprived of meaning in the case of LORF. Regarding coding strand diagnosis, we found no significant difference between LORF and UFM, which shows that in lower eukaryotes stop codon density is a strong coding frame identifier, even in a GC-rich genome such as that of
The average size of cORFs detected by UFM naturally increased with the cutoff threshold and was between 350 and 450 bp at the 95% success rate level, corresponding to a cutoff of 200 bp. The columns in Table 2 corresponding to “Bg≤BgBlst”, “End> EdBlst”, “PhsBlst” and “Bg&Phs” show the progression of the error. The order of these conditions in terms of decreasing success rates is as follows: “End> EdBlst” > “Bg≤BgBlst” > “PhsBlst” > “Bg&Phs”. The results show that incorrect ORFs tended to begin within homologous regions and extend over their corresponding in-frame cORFs. As would be expected, the strongest measure among was “PhsBlst”, which reports ORFs that are in the same frame as the homologous region (considered to be the true positives). However, an ORF could be located in the same frame as the homologous region, but not overlapping it. “Bg&Phs” shows that this event tends to disappear at the 95% level of consistency. We observed that the conditions “Bg&Phs” and “End>EdBlst” were redundant to the others and therefore not necessary (data not shown).
Coding Frame Classification when the Coding Strand is Unknown
In cases where the coding strand is unknown, the coding frame must be chosen from among six possibilities. The success rate of coding strand detection in such cases is given in Table 3. The trends observed in this table are similar to those of Table 2, except that the introduction of a variable corresponding to this strand introduces one additional degree of freedom. Consequently, the 95% consistency is achieved at a larger ORF size, typically 300 bp for higher eukaryotes and 150 (
Success rate of coding ORF diagnosis among the six frames of ESTs of

Sp: species name.
Regarding the evaluation of the success rate of coding strand diagnosis, introducing one degree of freedom in the strand increases the probability of obtaining an ORF in the same phase as the homologous region, but on the opposite strand. To filter this type of event out, it is necessary to score the proportion corresponding to the condition that the two events “Bg&Phs” and having the cORF on the “+” strand (that of the region homologous to PDB, by definition) occur together, which we denoted “Bg&Phs&Strd” in column 12 of Table 3. Based on comparison of Tables 2 and 3, it is obvious that the main source of error in GC-rich species produced by LORF is from diagnosis of the coding frame on the “–” strand when it is actually on the “+” strand. This is because the largest ORF is on the “–” strand in a significant proportion of the coding DNA in these species.
We found the consistency of the 200–300 bp cutoff for cORF to be acceptable because 93%–95% of the coding sequences of higher eukaryotes (taking

Size distribution of non-redundant coding sequences (GenBank) of
Since a success rate of ≥95% true positives corresponding to a sensitivity of >97% is obtained with UFM for ORFs ≥ 300 bp in ESTs of higher eukaryotes, it begs the question of what the success rate would be when the coding status is unknown, as is the case when BLAST homologies to PDB and/or other databases are not available.
Classifying cORFs in Rice ESTs
When the UFM classification was tested on ATG-Stop cORFs (the ATG-3n-Stop coding ORFs diagnosed by UFM) from rice EST contigs (n = 16,533), we did not find any consistent difference between the profiles of the observed (plain lines in Fig. 3) and expected frequencies (dashed lines in Fig. 3) for GC3 (Fig. 3A) or GC2 vs. GC3 (Fig. 4A). The histogram (GC3) and scatter plots (GC2 vs. GC3) actually match those found using the CDS sample from TIGR, corrected by AMI filtering for false positives.
19
The analysis of ATG-Stop cORFs from singlets (n = 8,068) revealed the same pattern for GC3 (Fig. 3B) and GC2 vs. GC3 (Fig. 4B), though with some possible false positives being observed in the compositional range below GC3 = 30%. Interestingly, the sum of cORFs (n = 24,600) from contigs (n = 16,533) and singlets (n = 8,068) is close to the reported gene number of approximately 25,000 for

GC3 distribution of ATG-Stop cORFs (>300 bp) in the rice transcriptome. (Panel

Scatter plots of GC3 vs. GC2 of ATG-Stop cORFs (>300 bp) in the rice transcriptome. (Panel
Prior Knowledge and cORF Classification in Human ESTs
In the case of the humans, we found that the GC3 of mRNA sequences of Fedorov's group17,18 calculated in reference with their first bases (Fig. 5A, thin line) is clearly out of frame. The GC3 calculated from their cORFs obtained with UFM (Fig. 5A and B, bold line) matched those of CDSs from that group (Fig. 5B, thin line) and from Zoubak et al 15 (Fig. 5B, dashed line). Consequently, most of the mRNAs from Fedorov's group17,18 are out of coding frame but the gene annotation is correct. The framing of mRNA by UFM (ATG-Stop ORF) is also correct. Similarly, the mismatch between the distributions of CDSs from the original dataset of Fedorov's group17,18 and the same dataset after filtration for homology with PDB sequences was only ~2% (Fig. 6A). The mismatch between the distributions of CDSs from Fedorov's dataset17,18 after filtration for homology with PDB sequences and that of Zoubak et al 15 was in the same range, which is reassuring given the much poorer knowledge of the human genome at that time (Fig. 6B).

The GC3 distribution in the reference dataset of human coding sequences. (Panel
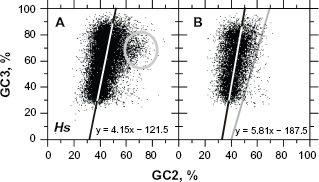
Scatter plotting of data is a powerful method for determining outliers that may be false positives. The scatter plots presented in Figure 6 show that the mismatch between the two GC3 distributions of Figure 5C is actually due to a small number of sequences within the gray circle in Figure 6A. These sequences are not present in the scatter plot shown in Figure 6B, which suggests that they are false positives because the plot in Figure 6B is based on sequences with undisputable experimental evidence (protein crystallization) and almost no dots are found on the right side of the line y = 3.33x – 130 (Fig. 6B). This line does not participate in the UFM classification process; it is only used as a guideline to facilitate plot-to-plot comparisons.
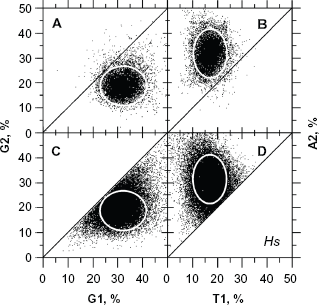
Considering the intron dataset of Fedorov's groups as representative of non-CDSs (ie, true negatives), as well as the dataset of curated CDSs from the same group that are homologous to PDB (ie, true positives) as representative of human CDSs, we determined that the classification threshold τ that generates the same rates (~11%) of false positives and false negatives (Fig. 7) took a value of 3.5 in humans when UFM was used without a posteriori filtering. Because A2 > T1 (Fig. 8A) and G1 > G2 (Fig. 8B) are generally true in the coding frame, a posteriori filtering under these conditions and a setting of τ ≥ 2 (Fig. 8C and D) decreased the rate of false positives, with no significant alteration of sensitivity (Fig. 9). However, it did not completely eliminate the false positives (as shown by the dots outside of the white ellipses and close to the diagonal in Fig. 8C and D), which is not surprising given the small difference in composition between CDSs and contiguous non-coding sequences. 21 Filtering out these false positives should only be considered on a case-to-case basis because it would affect the universality of UFM seeing as it would affect its applicability to other species without previous knowledge. The exact position and geometry of these ellipses actually change slightly according to the average GC level of the species under consideration (data not shown).

Histograms of UFM score in human introns (n = 10,348) and CDSs (n = 10,892). About 40% of introns (thin line, n = 3,346) gave UFM values larger than one.

Histograms of UFM score in human introns and CDSs with
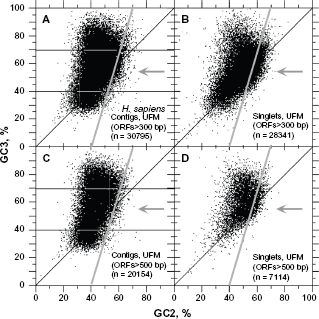
When the ATG-Stop ORFs were extracted by UFM from the contigs (n = 57,374) in the entire GenBank set of human ESTs (n=7,109,612), we found that their GC3 frequency was higher than the GC3 of CDSs on the GC-poor side of the reference distributions. When we filtered out the sequences shorter than 500 bp, the profiles of GC3 of ATG-Stop ORFs tended to match that of the reference dataset (Fig. 10C), suggesting that ~5% of false positives (the difference between the plain lines in Fig. 10A and C) fall in the range of 40% ≤ GC3 ≤ 70% for ORFs shorter than 500 bp. The difference between the plots shown in Figure 10A and C did not increase when filtering out ORFs larger than 500 bp (data not shown), suggesting that the cORFs in ~8% of the area (gray area) between thin and dashed lines in Figure 10C should indeed be considered as true positives and that the higher frequencies in the range of 40% < GC3 < 70% found for ATG-Stop ORFs compared to the reference distribution occurs simply because some GC-rich genes are expressed at lower rates than other genes. However, it is also true that for cORFs larger than 500 bp, a proportion of the cORFs outside the areas corresponding to the white ellipses in Figure 8C and D remain unchanged (data not shown), which demonstrates some of the cORFs in the 8% area (Fig. 10C) are indeed false positives. Figure 10B and D show that the ATG-Stop ORFs are much larger and that their proportion remains considerable in the 40% ≤ GC3 ≤ 70% interval of singlets, even at sizes larger than 500 bp. The fact that the frequency of ATG-Stop ORFs in singlets decreased more rapidly in the 50% < GC3 < 65% interval than in the 65% < GC3 < 75% interval (when only considering ORFs larger than 500 bp) is consistent with the hypothesis that they come from pseudogenes that are still expressed at very low levels.

GC3 distribution of ATG-Stop ORFs in human transcriptome compared to the genomic reference. ATG-Stop ORFs larger than 300 bp from contigs are more frequent in the 40% ≤ GC3 ≤ 65% interval compared to the reference by –13% (
The GC3 interval where we observed the most significant difference between the profiles shown in Figure 10A and C is marked by the gray arrows in Figure 11A and C. It is clearly visible from the lower density of dots in Figure 11C compared to Figure 11A in the 40% ≤ GC3 ≤ 70% range around GC2 = 55%, delimited by the line y = 3.33x – 130. Counting ~13% false positives in the 40% ≤ GC3 ≤ 70% interval, the number of human genes would be approximately 26,000 (47% of human EST contigs). However, it is impossible to calculate the proportion of singlets that could contribute to the gene number given the high noise level. Figure 11D suggests that at least 3,000 singlet cORFs could be true positives. Thus, the final expected number of true positive cORFs would be approximately 25,000–30,000, which is in agreement with the current view of the gene number in humans.
Scatter plot of GC3 vs. GC2 of ATG-Stop cORFs extracted with UFM from the human transcriptome. The gray arrow indicates the ATG-Stop ORFs from contigs larger than 300 bp that are more frequent in the 40% ≤ GC3 ≤ 70% interval and delimited by the line y = 3.33x – 130 compared to the reference (
In comparison with rice, the higher discrepancy between the profiles of ATG-Stop ORF distributions in contigs and singlets for GC3 histogram, as well as for GC2 vs. GC3 scatter plots found in humans, suggests a higher rate of genetic load accumulation in humans than in rice. This interpretation is supported by the plot shown in Figure 12, where genetic load accumulation would be higher in maize than in rice. This is in fact expected based on the higher rate of transposon and retrotransposon activity in maize than in rice.

Scatter plot of GC3 vs. GC2 of ATG-Stop cORFs extracted with UFM in maize transcriptome. The relationship of GC3 vs. GC2 from contigs is plotted in (
Searching for Functions in ORFs with a Purine Bias that are Apparently False Coding
In humans, a substantial proportion (n = 6,221) of ESTs from singlets (28,341, with nucleotides denoted as N, B, H, K, W, S, Y, and R) were of rather low quality, which make these sequences unique and hampers their assembly with the existing contigs. However, among the 28,341 singlet sequences, 22,120 did not contain an “N”, but 17,159 were <500 bp, which is a proportion that is too large (17,159/22,120 = 77.6%) to be explained by the low sequence quality (21.9%) of the sample. These sequences, which tended to be associated with the highest GC2 levels (GC2 > 50%, Fig. 11B and D), were also those where

The GC3 distribution of ATG-Stop cORFs from contigs homologous (BLASTn, E ≤ 0.0001) to

The GC3 vs. GC2 scatter plot of ATG-Stop cORFs from contigs homologous (BLASTn, E ≤ 0.0001) to
Testing MHC CDSs as another source of sequences with a possible bias, we found that these genes are indeed rich in GC2 (>40%), as observed in Figure 15A and B. However, the sample tested was out of the coding frame (ellipse of Fig. 15A) and needed to be analyzed using UFM to obtain a more reliable GC3 vs. GC2 relationship (Fig. 15B). We found that all ATG-Stop ORFs among human EST contigs homologous to MHC CDSs (BLASTn, E ≤ 0.0001) showed a GC3 content in the same range as that of GC2, suggesting their transformation in pseudogenes (Fig. 15C).
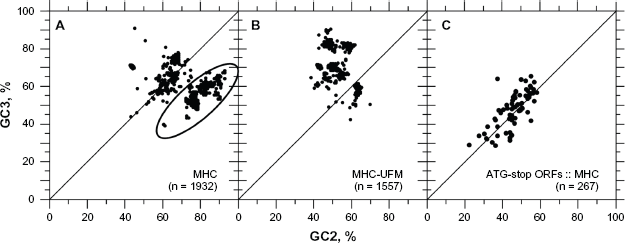
Scatter plot of GC3 vs. GC2 for MHC coding sequences. The sample of MHC coding sequences (CDS) retrieved from GenBank was found to be out of frame (black ellipse). In about half of the cases the frame + 1 has been confused with frame –3 (dots within the ellipse of Panel
Because the sequences from the singlet sample are not expressed more than one time among 7,109,612 ESTs (18,444/7,109,612 = 0.26%), it is not justified to consider them as true positives. This finding shows that the samples based on the distribution of ATG-Stop ORFs among contigs are more reliable than those based on ORFs in singlets, as the latter can be contaminated by noisy sequences.
At least some of the approximately 5% of putative false positives delimited by the line y = 3.33x – 130 in the 40% ≤ GC3 ≤ 70% range (n = 1,342) could have some biological significance, as they are expressed more than one time. By comparison to Gene Ontology (GO), we found 601 (45%) homologous sequences using BLASTx (Blast2GO, E ≤ 0.000001) against the
These annotations could be divided into three groups. The first group consists of cellular components (Fig. 16A), with eight non-redundant groups (n = 413) on the sixth GO level, ie, organelles (66%), nuclear parts (10%), cytosol (7%), nucleolus (6%), nucleoplasm (5%), ribosome (2%), cytoplasmic vesicle (3%), and cytoskeletal parts (2%). The second group consists of biological processes (Fig. 16B), with nine non-redundant groups (n = 361) on the fifth GO level, ie, signal transduction (20%), cellular macromolecule biosynthetic processes (20%), nucleic acid metabolic processes (20%), cellular protein metabolic processes (16%), protein transport (7%), establishment of protein localization (7%), ion transport (4%), regulation of cellular component size (3%), and regulation of macromolecule metabolic processes (1%). The third group consists of molecular functions (Fig. 16C), with twelve only slightly redundant groups (n = 446) on the third GO level, ie, protein binding (43%), hydrolase activity (12%), nucleotide binding (11%), transferase activity (10%), signal transducer activity (9%), transcription factor activity (4%), ion binding (3%), lipid binding (2%), carbohydrate binding (2%), chromatin binding (2%), and transmembrane transporter activity (1%), which means that ~80% seem to be involved in binding activities.

Annotation of the ATG-Stop cORFs extracted with UFM in the 45 < GC3 < 65 vs. 50 < GC2 < 60 interval of the human EST contigs. Annotations were obtained by comparing cORFs with
Discussion
UFM is a type of decision tree
22
in which a candidate coding ORF (cORF) flows across a sequence of tests involving less than 20 variables addressing objective criteria (eg, purine bias and stop codon frequency) that are the first determinants of coding DNA features. We showed above that UFM is a convenient tool for the extraction of cORFs from the transcriptome of any eukaryote. Considering the cORF classification among the six frames of CDSs homologous to the protein sequences of PDB, UFM provides an approximately 95% success rate in cORF diagnosis for approximately 95% of coding sequences (CDS) ≥300 bp in the case of higher eukaryotes. The fact that the same performance is obtained even for lower size (typically 200 bp) in
The size threshold corresponding to a 95% success rate of cORF classification among ESTs can be lowered by 100 bp to 200 bp in higher eukaryotes and 100 bp in lower eukaryotes if the coding strand is known. This is the case when, for instance, the polyA tail is present or when cDNA are obtained through directional
UFM allows automatic transcriptome phenotype visualization in the compositional sense given to the genome phenotype by Bernardi. 23 That is, UFM allows the fast calculation of the distribution of CDSs according to GC3, in an impromptu manner, on the output of cDNA sequencing. Recording the number of cORFs assembled in each contig provides information regarding the level of expression of the sequenced genes. Mounting contigs from cORFs of ESTs has the side effect of measuring the level of expression just by counting ESTs per contig, for example, visualizing or picturing the transcriptome phenotype also allows the RNA-Seq to be performed.24,25
Due to the compositional transition that occurred in
When considering plants, we found that the GC3 vs. GC2 plots of ATG-Stop cORFs from contigs match that of the universal correlation. This strongly suggests that UFM can be used on the transcriptome of any plant species for extracting cORFs, without any additional knowledge or parametric tuning. This was true even in new cases, such as for genetic prospecting for exploration of biodiversity. The same plot for ATG-Stop cORFs obtained from singlets also matches the universal correlation, except in case of maize, where the plot is contaminated by noisy sequences in the ranges of 40% ≤ GC3 ≤ 70% and GC2 ≥ 50%. This phenomenon, which is not observed in rice, is much stronger in humans. This observation also suggests that a possible alternative to this problem is the transcriptome analysis of a species of the same family that is known to have a small genome. The analysis could then serve as a mold for discarding, by subtraction, the junk information from the larger genome. In parallel, it could be tempting to extend the definition of genetic load—ie, the aggregate of deleterious genes that are carried, mostly hidden, in the genome of a population and may be transmitted to descendants—to involve retrotransposon activity. In this perspective, the relative importance of noisy sequences in large genomes follows what may be expected concerning the dynamics of the genetic load, suggesting that these sequences are derived from pseudogenes, particularly those induced by transposition.
28
In humans, these sequences are expressed more than one time in some cases, which means that they potentially exhibit a biological role and have to be considered as true positive cORFs. We found that 45% of these sequences actually have a biological function in humans, with the functions relating to bonding activities in ~80% of cases. Interestingly, the compositional distribution of MHCs may very well partly justify such a hypothesis.
Conclusions
The UFM algorithm is expected to be suitable for preliminary transcriptome mining of any eukaryote, without prior knowledge of that species. It may also be considered as a tool in assisting the first steps of genome annotation for pure or mixed species samples. The very low level (close to the information content) of this algorithm based on objective and universal determinants of coding sequences (eg, stop codon density, purine bias, ORF size) makes it sensitive to false positive sequences that mimic the purine bias of typical protein gene, such as pseudogenes, transposons, and retrotransposons. Fortunately, these sequences are expressed at low rates, which allows them to be reasonably discriminated via scoring their relative rate of expression or, more simply, via contig assembly. A corollary of this is that their impact is lower (or null) in small genomes 37 that optimized their removal as part of an evolutionary strategy. This image is similar to a molecular representation of the concept of genetic load. Given the high genetic load that eventually accumulates in higher eukaryotes, transcriptome mining appears to be an obligate path toward genome annotation for proper filtering out of junk data.
Author Contributions
Conceived and designed the experiments: NC. Analysed the data: NC. Wrote the first draft of the manuscript: NC. Contributed to the writing of the manuscript: NC, DF. Agree with manuscript results and conclusions: NC, DF. Jointly developed the structure and arguments for the paper: NC, DF. Made critical revisions and approved final version: NC and DF. All authors reviewed and approved of the final manuscript.
Funding
This research was supported by the Brazilian agencies FIOCRUZ/CDTS and CAPES, which provided a research fellowship to N. Carels. We thank
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
Pat. Req. PI1001614-7.
Footnotes
Acknowledgements
We thank