
Abstract
Pulmonary arterial hypertension (PAH) is a chronic, progressive disease of the pulmonary vasculature with a high morbidity and mortality. Its pathobiology involves at least three interacting pathways – prostacyclin (PGI2), endothelin, and nitric oxide (NO). Current treatments target these three pathways utilizing PGI2 and its analogs, endothelin receptor antagonists, and phosphodiesterase type-5 (PDE-5) inhibitors. Inhaled nitric oxide (iNO) is approved for the treatment of hypoxic respiratory failure associated with pulmonary hypertension in term/near-term neonates. As a selective pulmonary vasodilator, iNO can acutely decrease pulmonary artery pressure and pulmonary vascular resistance without affecting cardiac index or systemic vascular resistance. In addition to delivery via the endotracheal tube, iNO can also be administered as continuous inhalation via a facemask or a pulsed nasal delivery. Consistent with a deficiency in endogenously produced NO, long-term pulsed iNO dosing appears to favorably affect hemodynamics in PAH patients, observations that appear to correlate with benefit in uncontrolled settings. Clinical studies and case reports involving patients receiving long-term continuous pulsed iNO have shown minimal risk in terms of adverse events, changes in methemoglobin levels, and detectable exhaled or ambient NO or NO2. Advances in gas delivery technology and strategies to optimize iNO dosing may enable broad-scale application to long-term treatment of chronic diseases such as PAH.
Keywords
Pulmonary arterial hypertension (PAH) is a chronic, progressive disease of the pulmonary vasculature resulting in right ventricular failure and death, if untreated.[1,2] PAH is defined by the following: a resting mean pulmonary arterial pressure (mPAP) ≥25 mmHg; pulmonary capillary wedge pressure or left ventricular end diastolic pressure ≤15 mmHg; and pulmonary vascular resistance (PVR) ≥3 Wood units.[2] PAH can be idiopathic, heritable, or associated with other conditions, such as connective tissue diseases (CTDs).[3,4]
The prevalence of PAH was estimated as 26–52 cases per million from the Scottish epidemiological study; a more conservative lower-bound estimate from the French PAH Registry reports 5–25 cases per million.[5,6] Prevalence is greater in high-risk groups, such as patients with CTDs, congenital heart disease (repaired and unrepaired), human immunodeficiency virus, and portal hypertension.[7–9]
Pulmonary arterial hypertension: Mortality and unmet medical need
The mortality with PAH remains high despite treatment advances over the past several decades. In the 1980s, the 5-year survival rate for idiopathic PAH (IPAH; formerly termed “primary pulmonary hypertension”) was 34% in the National Institutes of Health (NIH) Registry; although 5-year survival has increased to ~60% using currently available drugs, the mortality remains unacceptable.[4] Patients in the NIH registry in the 1980s were treated with the conventional therapy available at the time, including diuretics, digoxin, supplemental oxygen, warfarin, and calcium channel blockers (if clinically indicated).[4] Prior to 1995, no drugs were approved for PAH. However, there are currently eight drugs approved for the treatment of PAH: intravenous (IV) epoprostenol, IV/subcutaneous (SC) treprostinil, inhaled treprostinil, inhaled iloprost, oral bosentan, oral ambrisentan, oral sildenafil, and oral tadalafil. A meta-analysis of all randomized, controlled PAH trials published through 2008 suggested that with these available PAH-specific treatments, mortality has decreased 43% (RR: 0.57; 95% CI: 0.35–0.92;
Patients with PAH also report severe impairment of health-related quality of life (HRQOL), including poor general and emotional health, and impaired physical functioning.[9] These impairments to HRQOL with PAH are comparable and not infrequently greater than those reported in patients with severely debilitating conditions such as spinal cord injury or cancers unresponsive to therapy.[9] Improvement in HRQOL scores has been reported (e.g., increased exercise capacity and physical functioning) utilizing the currently available PAH-specific drugs.[11–13]
Pathobiology
The postulated pathobiology of PAH involves interactions between the prostacyclin (PGI2), endothelin (ET-1), and nitric oxide (NO) pathways, in addition to a host of other pathways (Fig. 1).[2,14–16] Specific mechanisms responsible for the development and progression of PAH include the following: reduced PGI2 synthase; increased ET-1 expression; decreased NO synthase; elevated plasma levels and low platelet 5-hydroxytryptamine levels; downregulation of potassium channels of pulmonary vascular smooth muscle cells; activity of autoantibodies and proinflammatory cytokines; and prothrombotic states arising from endothelial, coagulation, and fibrinolytic cascade/platelet dysfunction.[16] These changes give rise to a complex process of pathobiologic changes in the pulmonary vascular bed, including endothelial dysfunction, vasoconstriction, vascular remodeling, and in situ thrombosis.[2]
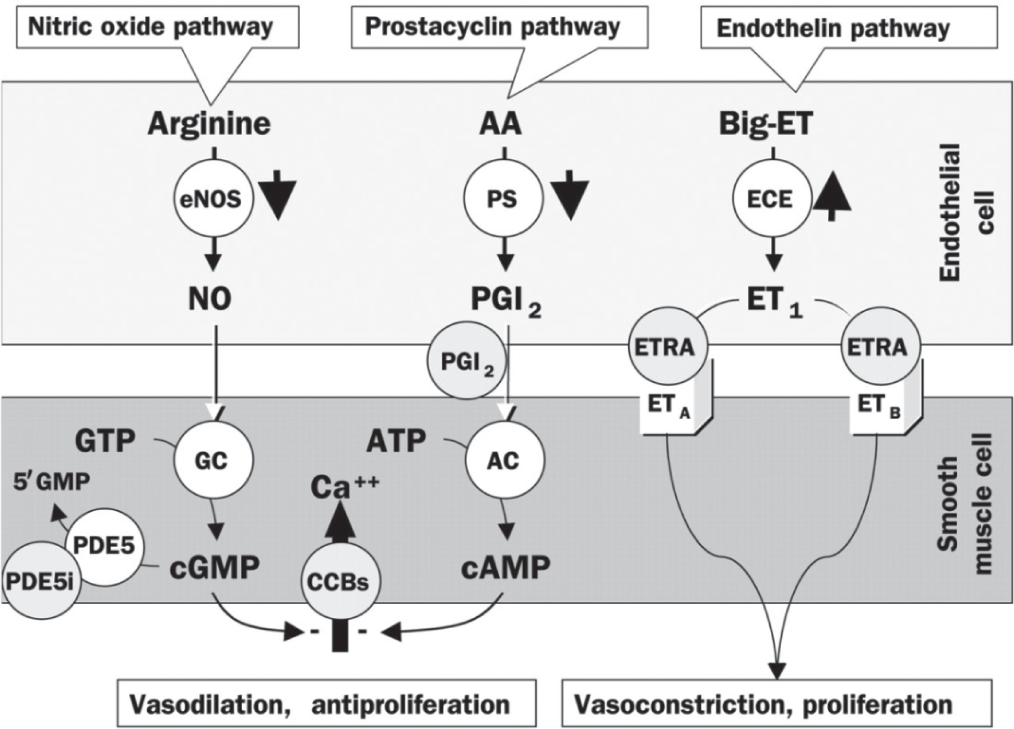
Pathways involved in the development and maintenance of pulmonary arterial hypertension. AA: arachidonic acid; ET: endothelin; eNOS: endothelial NO synthase; PS: prostacyclin synthase; ECE: endothelin-converting enzyme; PGI2: prostaglandin I2 (prostacyclin); ETRA: endothelin receptor agonist; GTP: guanylate triphosphate; GC: guanylate cyclase; ATP: adenosine triphosphate; AC: adenylyl cyclase; CCB: calcium channel blocker; cGMP: cyclic guanylate monophosphate; cAMP: cyclic adenosine monophosphate; PDE5: phosphodiesterase-5; PDE5i: PDE5 inhibitor. Reprinted from The Lancet, Vol. 358, Jocelyn Dupuis, Endothelin-receptor antagonists in pulmonary hypertension, pages no. 1113–1114, Copyright (2001), with permission from Elsevier[15] and with permission from Mayo Clinic Proceedings, Volume 84, Michael D. McGoon and Garvan C. Kane, pulmonary hypertension: diagnosis and management, pp 191–207, Copyright Mayo Foundation for Medical Education and Research (2009).[16]
Pharmacologic targets of currently approved treatments for PAH
Current PAH treatment approaches include PGI2 and its analogs, ET-1 receptor antagonists (ERAs), and phosphodiesterase type-5 (PDE-5) inhibitors.[17] Combination trials have demonstrated additive and/or synergistic benefit by targeting more than one pathway.[17] Prostanoid monotherapy (epoprostenol, treprostinil, and iloprost) improves symptoms, exercise capacity, and hemodynamics.[17] Increased survival was also demonstrated in IPAH/heritable PAH (HPAH) with IV epoprostenol. However, common side effects with prostanoids include headache, flushing, nausea, jaw pain, diarrhea, skin rash, and musculoskeletal pain. Treatment with PGI2 and its analogs often requires continuous intravenous parenteral infusion, which can cause blood stream infections and/or thromboembolic events that can be life threatening.[2]
Endothelin-1 exerts vasoconstrictor and mitogenic effects, whereas ERAs (i.e., bosentan and ambrisentan) improve exercise capacity, functional class, and hemodynamics.[2,8] Adverse effects include acute hepatotoxicity, anemia, and fluid retention. Additionally, ERAs may cause testicular atrophy and male infertility. Use of bosentan requires monthly liver function tests and two modes of birth control, as it has been shown to cause severe fetal toxicity in animal studies.[2,18]
In three randomized trials, the PDE-5 inhibitors sildenafil and tadalafil improved exercise capacity and hemodynamics (either as monotherapy or as add-on therapy).[8,11,17] Both agents cause pulmonary vasodilation.[8] Side effects include headache, flushing, and dyspepsia and are generally related to systemic vasodilation. Epistaxis has also been reported with sildenafil use in PAH.[8,11] Prostacyclin analogs, ERAs, and PDE-5 inhibitors are the mainstays of current PAH treatment; however, all have systemic effects in addition to their pulmonary effects that can cause untoward side effects.[19] An optimal agent for PAH therapy remains to be identified.[17]
Inhaled nitric oxide
Inhaled nitric oxide (iNO) is a selective pulmonary vasodilator that can acutely decrease pulmonary artery pressure (PAP) and pulmonary vascular resistance (PVR) in neonates with hypoxic respiratory failure associated with pulmonary hypertension.[20] Nitric oxide regulates vascular smooth muscle tone and increases blood flow to regions of the lungs with normal ventilation/perfusion ratios by dilating pulmonary vessels in better-ventilated areas.[21] After inhalation, NO is absorbed systemically, with the majority of NO traversing the pulmonary capillary bed and combining with 60–100% oxygen-saturated hemoglobin.[20] The effect of iNO is localized to the lung, as once absorbed iNO is rapidly oxidized by hemoglobin to form nitrite, which interacts with oxyhemoglobin, leading to the formation of nitrate and methemoglobin (metHb).[20,22] This metabolic production of metHb is a potential toxic effect of iNO treatment. While doses <100 ppm most often result in insignificant metHb levels in adults and children, methemoglobinemia has been reported with 80 ppm when exposure was >18 hours.[23]
Inhaled NO is currently indicated for the treatment of term/near-term neonates (>34 weeks gestation) with hypoxic respiratory failure associated with pulmonary hypertension (PH). The recommended dose is 20 ppm delivered via constant concentration during inspiration for up to 14 days or until hypoxia has resolved.[20]
Inhaled NO has also been used as an agent for acute vasodilator testing (AVT) as part of the evaluation of PAH patients; doses of 20–80 ppm for 5–10 minutes are typically used.[2,24] Detecting an acute response with AVT is useful in selecting patients who should be considered for initial treatment with high-dose oral chronic calcium channel blockade; AVT response may also be helpful in predicting long-term prognosis with medical therapy and following surgical interventions, such as heart or heart–lung transplantation.[24] Administered as continuous inhalation via face mask, iNO can selectively decrease PAP and PVR without reducing cardiac index or systemic vascular resistance.[24] Inhaled NO has also been used in other contexts, such as perioperatively for cardiac surgery,[25–33] right heart failure after insertion of the left ventricular assist device,[34–37] cardiogenic shock due to right ventricle myocardial infarction,[38] and pulmonary ischemia-reperfusion injury.[39–44]
Because the pulmonary vasodilator effects of NO are transient, it is administered continuously during inspiration, with careful monitoring of NO and NO2 concentrations.[20] Nitric oxide gas can be safely administered in both intubated and nonintubated patients.[45] The pulmonary selectivity of iNO may render it useful as an adjunct to other therapies that are dose limited by their systemic effects.
Inhaled NO has also been administered long term via pulsed nasal delivery (ml/breath/h) in clinical trials; this method has been studied for continuous long-term outpatient as well as short-term inpatient treatment (Fig. 2).[46–49] This ambulatory administration method delivers a set, pulsed volume of NO at the beginning of each breath via a nasal cannula connected through a NO demand valve to a cylinder of up to 200 ppm NO in N2.[46–48] Both the continuous face mask and pulsed delivery via nasal cannula have comparable hemodynamic effects.[50] A potential theoretical advantage of iNO, in contrast to IV vasodilators, is its pulmonary selectivity (due to rapid hemoglobin-mediated inactivation).[22] Although prostanoids administered via inhalation appear to have less ventilation–perfusion mismatching than when administered intravenously/ subcutaneously or orally, some degree of ventilation–perfusion mismatching persists; in addition, systemic spillover can result in untoward systemic effects.[51–53]
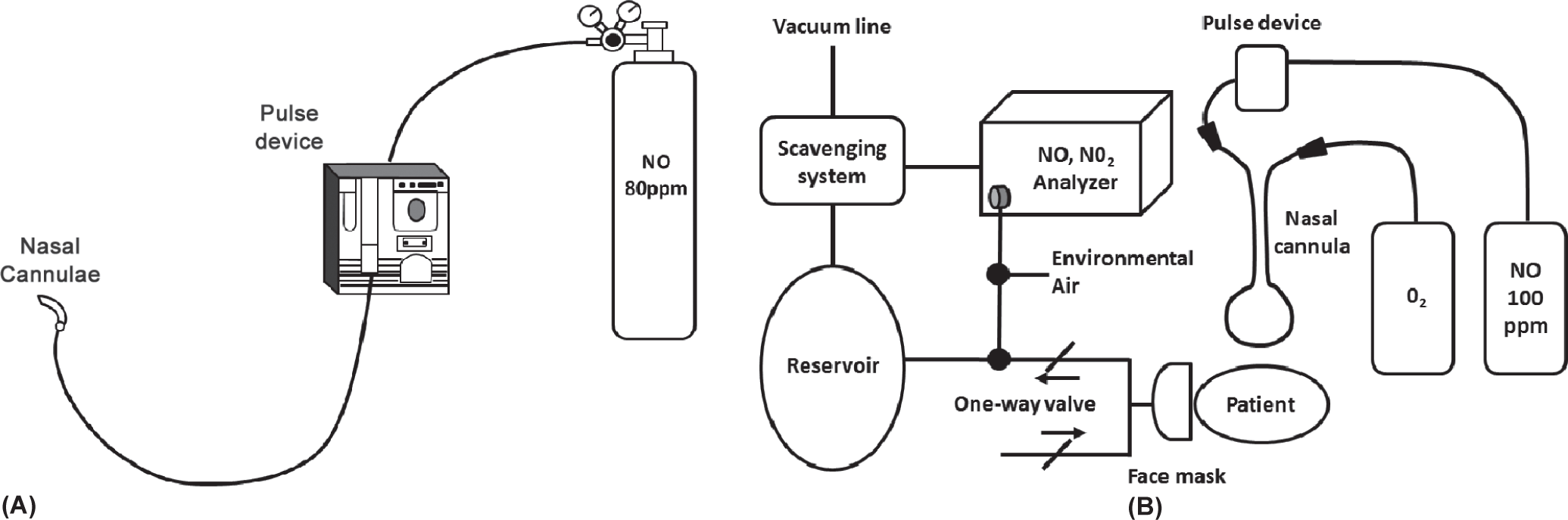
Examples of pulsed inhaled nitric oxide delivery systems used in clinical studies:
CLINICAL APPLICATION OF INHALED NITRIC OXIDE AS LONG-TERM TREATMENT FOR PAH
Long-term (>1 month) pulsed iNO dosing appears to favorably affect pulmonary hemodynamics findings[46–48,54,55] which, with other types of therapy, appear to correlate with benefit (Table 1).
Inhaled long-term nitric oxide use for treatment of pulmonary arterial hypertension
In a study of eight patients with IPAH, Channick et al. reported decreased mean PAP (mPAP), mean right atrial pressure (mRAP), and PVR (
Ivy et al. also reported that in 26 children and young adults with PAH (short-term therapy, n=24; long-term therapy, n=2) constant concentration and pulsed delivery of NO (via nasal cannula) were equally effective in decreasing PAP and PVR (
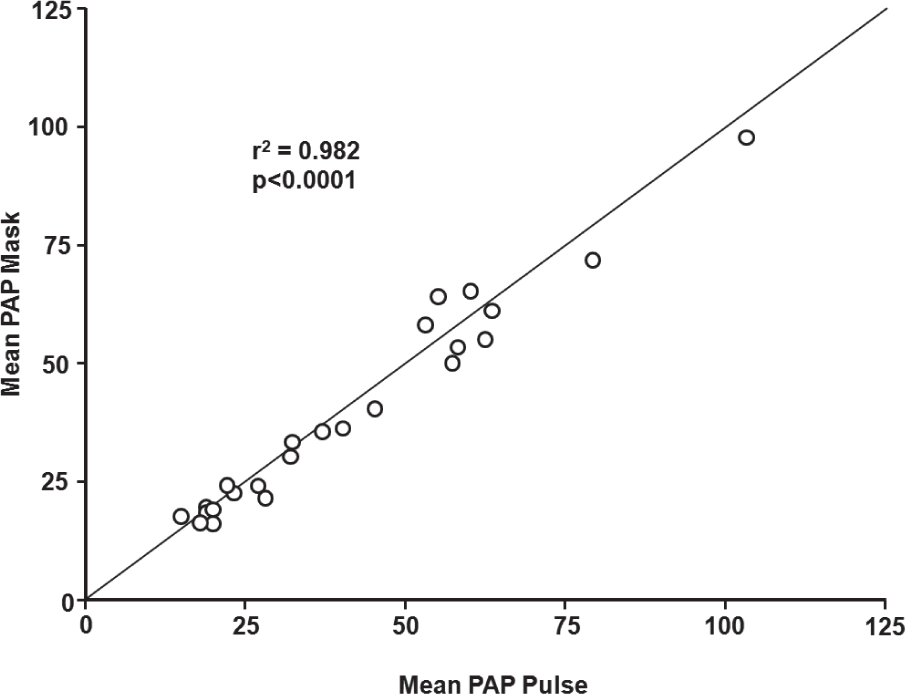
Correlation between mean pulmonary arterial pressure during mask delivery and pulsed nasal nitric oxide delivery. PAP: pulmonary artery pressure. Reprinted from The American Journal of Cardiology, Vol 92, D. Dunbar Ivy, Donna Parker, Aimee Doran, Donna Parker, John P. Kinsella, and Steven H. Abman, acute hemodynamic effects and home therapy using a novel pulsed nasal nitric oxide delivery system in children and young adults with pulmonary hypertension, pages no. 886–890, Copyright (2003), with permission from Excerpta Medica, Inc.[46]
Long-term treatment with pulsed iNO was evaluated in 11 patients (7 with PAH and 4 with chronic thromboembolic PH) in an uncontrolled, open-label study. The study design included the addition of PDE-5 inhibitor (dipyridamole or sildenafil) for clinical worsening; this was suggested as a means to “stabilize and potentiate the effects of iNO” and to “potentially serve as rescue therapy in severe PH” (Table 1).[47] After 1 month of an ambulatory iNO system via nasal cannula, patients had an improvement in World Health Organization functional class concomitant with improvements in 6-minute walking distance (
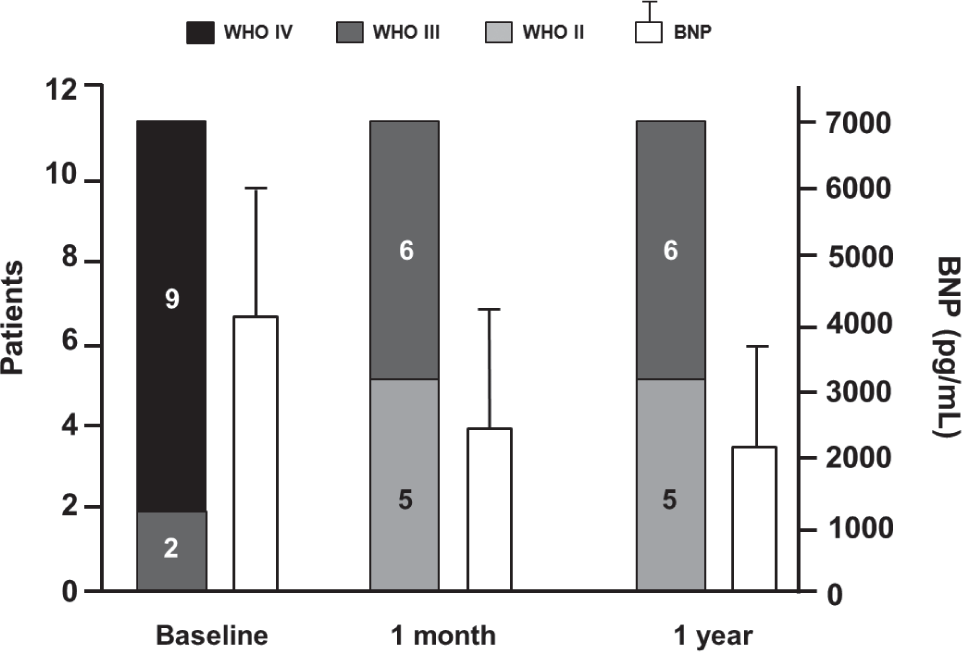
World Health Organization functional class and brain natriuretic peptide levels (mean±SD) at baseline compared with 1 month and 1 year after onset of iNO treatment. *In Patients 1 and 2, the measure was taken at 6 months. BNP: brain natriuretic peptide. Reprinted from The Journal of Heart and Lung Transplantation, Vol 27, Gregorio Miguel Pérez-Peñate, Gabriel Juliá-Serdà, Nazario Ojeda-Betancort, Antonio García-Quintana, Juan Pulido-Duque, Aurelio Rodríguez-Pérez, Pedro Cabrera-Navarro, Miguel Angel Gómez-Sánchez, Long-term inhaled nitric oxide plus phosphodiesterase 5 inhibitors for severe pulmonary hypertension, Pages No. 1326–1332, Copyright (2008), with permission from the International Society for Heart and Lung Transplantation.[47]
Two case reports have also examined long-term iNO administration in PAH patients, including its use as a “bridge to heart-lung or lung transplantation” (Table 1). A 40-year-old woman presented with end-stage IPAH and experienced severe dyspnea, right ventricular angina, oliguria, and syncope despite treatment with dopamine infusion and with prostacyclin. The patient then initiated treatment with pulsed iNO, initially via face mask and then transtracheal catheter, until she underwent heart–lung transplantation after 68 days of therapy.[55] The patient's condition appeared to stabilize on iNO treatment, although she had a hypotensive bradycardic event after 53 days, requiring reinitiation of intravenous prostacyclin. While iNO was administered, she was able to move about her room independently and participate in a physiotherapy exercise program. The explanted lungs revealed no evidence of NO toxicity.[55]
Another case reported the effects of 12 months of iNO administration in a 32-year-old man with IPAH (Table 1).[54] The patient presented with exertional dyspnea and anasarca, and was treated with long-term iNO monotherapy via an ambulatory system with nasal cannula. After 20 days, there was an improvement in dyspnea and gas exchange, and a resolution of the anasarca. After 12 months of continuous iNO, the patient remained clinically stable, with maintained hemodynamic improvement and no signs of toxicity or tachyphylaxis.[54]
Ivy et al. reported that short-term pulsed nasal delivery utilizing constant concentration was as effective in lowering PAP and PVR as mask delivery in the acute setting in eight children with PAH (Fig. 5).[50] Based on the results of this study, the authors concluded that the practicality of long-term iNO therapy via pulsed flow nasal delivery is potentially dependent on four factors: (1) maintenance of sufficient iNO delivery; (2) improvement of hemodynamic derangements by nasal cannula at low flow rates; (3) effective delivery of nasal NO with minimal release of gas into the environment; and (4) minimized consumption of NO gas.[50] Aside from the reports involving long-term use summarized in Table 1, the practicality of pulsed delivery of iNO for improvement in oxygenation with less NO consumption and less environmental contamination has been demonstrated in several other studies.[56–58]
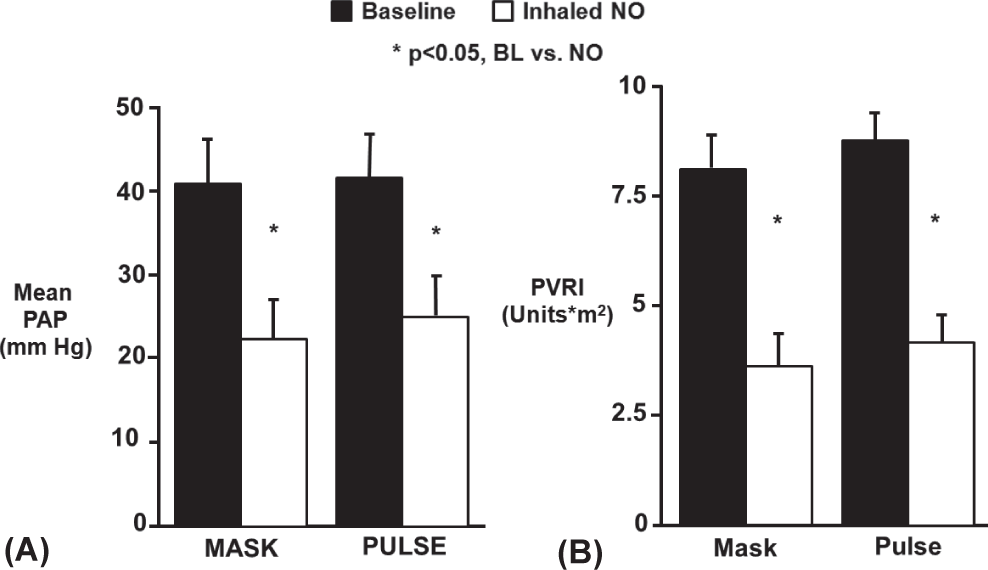
Delivery of inhaled NO by continuous mask or pulsed nasal cannula was equally effective in lowering mean pulmonary artery pressure
ROLES FOR LONG-TERM INHALED NITRIC OXIDE IN THE TREATMENT OF PULMONARY ARTERIAL HYPERTENSION
Potential uses of pulsed, long-term iNO treatment in PAH patients include the following: use as a bridge to transplantation; a means of deferring transplantation; and as an add-on therapy to currently approved PAH drugs[2,45] with potential additive or synergistic effects.[59,60] It is important to note that NO synthase 3 (NOS3) has been reported to be decreased in PAH patients; in uncontrolled observational studies, PAH has been associated with impaired NO release, at least in part, due to reduced expression of NOS3 in the vascular endothelium of pulmonary arterioles.[61] As a result, long-term administration of iNO may serve both as a selective pulmonary vasodilator and as NO replacement therapy, making it a logical choice for clinical evaluation as add-on therapy.
Safety considerations
A potential safety concern with iNO treatment is rebound PH upon its sudden discontinuation after longer-term (days) use[20,62]; this phenomenon is well known and has been well documented in neonates and in postoperative cardiac surgery patients. Such patients include cardiac transplant recipients, children undergoing surgery for congenital heart disease, and adults with mitral and/or aortic stenosis. Gradual weaning of iNO has been shown to minimize the potential for rebound PH in the acute ICU setting.[45] Davidson et al. presented a method to safely withdraw iNO in infants treated for hypoxic respiratory failure, recommending the gradual weaning of iNO down to 1 ppm prior to treatment discontinuation.[63] Further research has implicated the rapid degradation of smooth muscle intracellular cyclic guanosine monophosphate (cGMP) by local phosphodiesterases (PDEs) as a primary mechanism for this rebound effect. As a result, initial approaches focused on the use of dipyridamole, a PDE-5 inhibitor, as a means for reducing rebound PH after iNO withdrawal. Ivy et al. first demonstrated this concept in a prospective study of 23 children treated with iNO after surgery for congenital heart disease.[64] Later studies examined the role of sildenafil, another PDE-5 inhibitor, in the context of rebound PH, showing that its introduction prior to withdrawal of iNO resulted in facilitation of iNO weaning, as well as prevention/amelioration of rebound PH effects in infants and children with PH after congenital heart disease surgery, persistent PH of the newborn, and other abnormalities.[65–68] As with any approach, it is important to consider patient characteristics and treatment familiarity, availability, and contraindications, as well as optimal ventilation and supplemental vasodilators, when initiating treatment for rebound PH.[66]
A review of the published literature on long-term iNO dosing in PAH patients has not revealed any reports of rebound PH crises or associated symptoms (e.g., syncope, systemic arterial oxygen desaturation, systemic hypotension, bradycardia, or cardiac arrest).[46–48,54,55] It may be that more acute initial rise in PAP is associated with a greater likelihood and severity of a rebound effect occurring with acute iNO withdrawal. This may explain why the rebound phenomenon has been observed in the acute care setting (e.g., neonates with persistent pulmonary hypertension of the newborn and high risk postoperative cardiothoracic surgical patients) and not observed in the more chronic setting of PAH or chronic obstructive pulmonary disease.[69]
Cytotoxicity is another possible concern with iNO and its oxidized derivatives (principally NO2). Nitric oxide may be directly toxic to alveolar and vascular tissue; therefore, it has been proposed that NO be stored in combination with nitrogen and blended with oxygen at the time of administration to prevent oxidation to toxic products, in addition to maintaining NO2 levels <5 ppm.[23,70]
CONCLUSIONS AND FUTURE DIRECTIONS
In summary, uncontrolled observational studies of long-term use (>1 month) of continuous pulsed iNO (as monotherapy or as part of combination therapy) in a total of 14 patients with PAH across five studies[46–48,54,55] have reported no significant adverse events, no elevated metHb levels, and no detectable exhaled or ambient NO or NO2. In one study, a patient experienced three episodes of severe epistaxis over two years while on a combination of pulsed iNO and epoprostenol.[46] In a case report of a patient awaiting heart–lung transplantation, the patient experienced hypotensive bradycardia upon an attempt to wean from iNO therapy. In addition, a recurrence in hypotensive bradycardia resulted in the increase of iNO dose (40–106 ppm), followed by a decrease to 70 ppm (along with administration of bicarbonate and reintroduction of prostacyclin) after increasing metabolic acidosis.[55]
There is evidence that pulsed delivery may allow utilization of lower NO concentrations compared with continuous face mask administration, potentially minimizing the risk of associated adverse events as well as resulting in a more practical delivery system.[49]
The consensus on treatment for PAH encompasses numerous goals, the most important being to improve overall quality of life by decreasing symptoms while minimizing treatment-related side effects.[2] Additional goals include enhancing functional capacity, i.e., exercise capacity, improving hemodynamic derangements (lowering PVR and PAP, and normalizing RAP and CO), and preventing, if not reversing, disease progression. Finally, improving survival, although certainly desirable, is rarely an end point in trials examining PAH treatment.[2] The availability of novel treatments and the improvement in survival rates have allowed the goals of PAH therapy to expand from improving survival and preventing disease progression to also improving HRQOL.[71] Potential advances in long-term PAH treatment, such as ambulatory iNO administration, may allow for greater improvements in HRQOL. Pérez–Peñate et al. observed that ambulatory pulsed iNO treatment did not diminish quality of life beyond the consequences of the disease itself.[47] Eight of eleven patients who led a nonsedentary life were able to leave their home daily, with four returning to work while on long-term iNO therapy.
An ideal drug-device for long-term PAH treatment should emphasize portability and safety features for outpatient use. Advances in iNO gas delivery technology and strategies to optimize dosing should allow for randomized controlled trials of iNO and, hopefully, may lead to broad-scale application of iNO in the treatment of chronic diseases such as PAH.[45]
Footnotes
ACKNOWLEDGMENTS
The authors thank Michael Morren, RPh, of Peloton Advantage, LLC, for providing medical writing and editorial assistance, which was funded by Ikaria, Inc., during the preparation of this manuscript. No author received an honoraria or other form of financial support for the preparation of this manuscript.