
Abstract
The pulmonary vasculature comprises a complex network of branching arteries and veins all functioning to reoxygenate the blood for circulation around the body. The cell types of the pulmonary artery are able to respond to changes in oxygen tension in order to match ventilation to perfusion. Stem and progenitor cells in the pulmonary vasculature are also involved, be it in angiogenesis, endothelial dysfunction or formation of vascular lesions. Stem and progenitor cells may be circulating around the body, residing in the pulmonary artery wall or stimulated for release from a central niche like the bone marrow and home to the pulmonary vasculature along a chemotactic gradient. There may currently be some controversy over the pathogenic versus therapeutic roles of stem and progenitor cells and, indeed, it is likely both chains of evidence are correct due to the specific influence of the immediate environmental niche a progenitor cell may be in. Due to their great plasticity and a lack of specific markers for stem and progenitor cells, they can be difficult to precisely identify. This review discusses the methodological approaches used to validate the presence of and subtype of progenitors cells in the pulmonary vasculature while putting it in context of the current knowledge of the therapeutic and pathogenic roles for such progenitor cells.
Keywords
INTRODUCTION
The current exploitation of stem cells as a therapeutic approach and research tool is due to their extraordinary ability to both self renew through mitotic cell division and differentiate into a vast array specialized cell types.[1] Stem cells, as a broad terminology, reflect two distinct cell types: (1) embryonic stem cells (ESC), which are pluripotent, having the ability to both self renew indefinitely and differentiate into cells of all 3 germ layers (endoderm, ectoderm and mesoderm); and (2) adult stem cells, which have differentiated but retain some capacity to self-renewal and are more restricted in their potential to differentiate.[2,3] For example, some adult stem cells (or tissue specific stem cells) are capable of giving rise to several specialized cell types (multipotent stem cells) while others are limited to a single specialized cell type (unipotent stem cell).[4] The descendants of stem cells and representing the next level of differentiation are progenitor cells. These cells have lost the ability for self-renewal. Stem cells and progenitor cells exist in a hierarchical system gradually becoming more lineage restricted. This system has been most comprehensively studied in the hematopoietic system as highlighted in Fig. 1. This intricate hierarchy system exists to preserve a homeostatic repair and maintenance of the body, replenishing specialized cells and sustaining the routine cellular turnover in regenerative organs.[5] Adult stem and progenitor cells may be either circulating or resident in a particular tissue/organ system. Several are known to be present in the lung and pulmonary vasculature including endothelial progenitor cells (EPC), mesenchymal stem cells (MSC), and hematopoietic stem cells (HSC).[3] This review describes commonly used methods to identify adult stem and progenitor cells currently known to be present in the pulmonary vasculature. This review also provides a practical instruction of the methodological approaches used to study pathogenic and therapeutic role of stem/progenitor cells in pulmonary vascular disease.
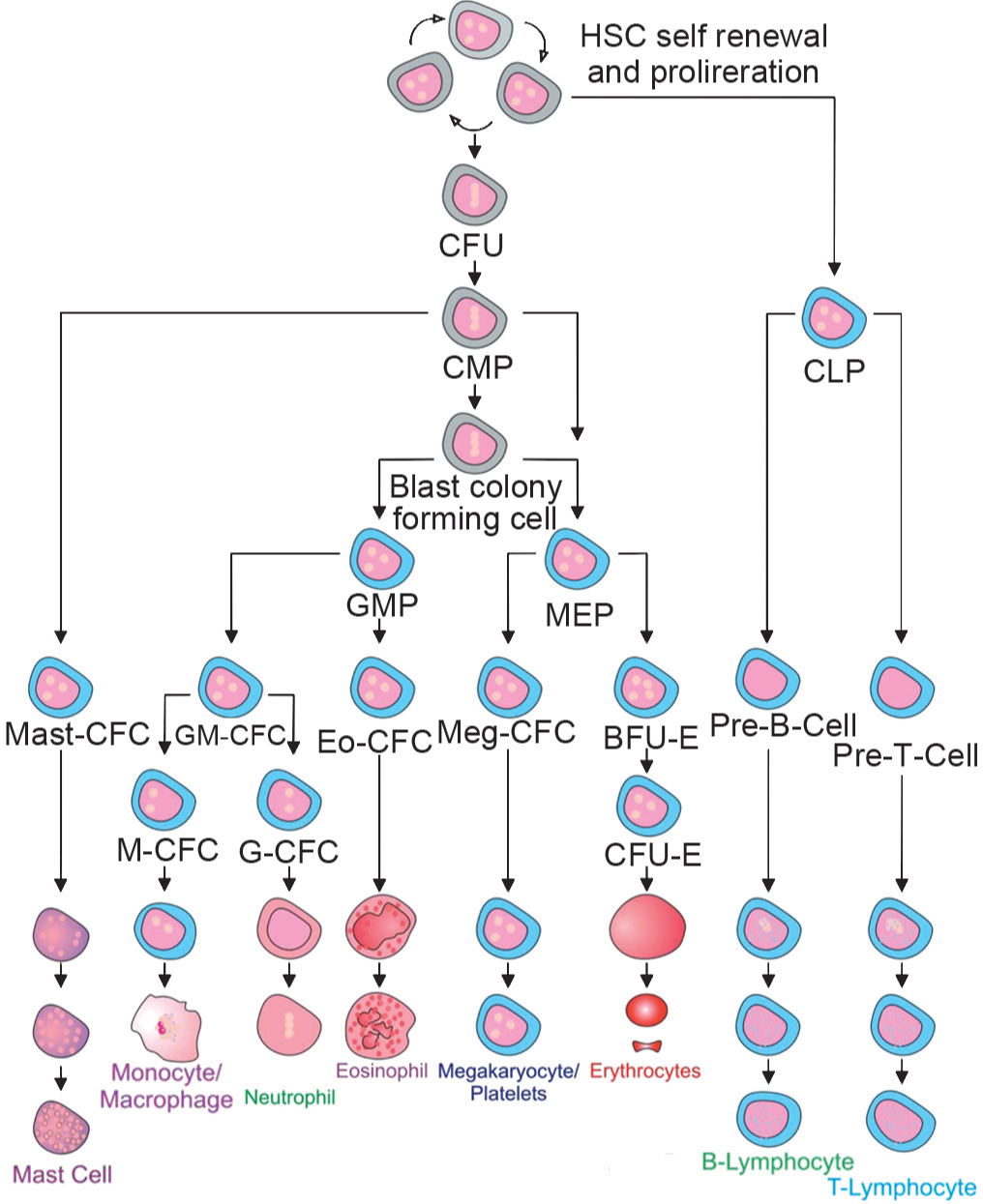
Hematopoietic stem cell hierarchy. Self-renewing HSC give rise to several multipotent progenitors (colony forming units (CFU), common myeloid progenitor (CMP) and common lymphoid progenitors (CLP)), which, in turn, produce oligopotent progenitors, unipotent progenitors and eventually fully differentiated cells. The CMP is able to produce granulocyte-macrophage progenitors (GMP) and megakaryocyte/erythrocyte progenitors (MEP) giving rise to monocyte/macrophages/granulocytes and megakaryocytes/platelets/erythrocytes, respectively. Erythoid burst forming unite (BFU-E) give rise to pro-erythroblast colony forming unit–erythroid (CFU-E) before erythrocytes are formed and the CLP gives rise to pre-B and pre-T cells which continue to mature into mature B and T lymphocytes. (Adapted with permission from reference 84).
DISCUSSION
Stem cells in the pulmonary vasculature
Paradoxically, stem cells may have both a therapeutic benefit and a pathogenic role in the pulmonary vasculature. Evidence to date suggests that hematopoietic stem cells likely have a pathogenic role being elevated in perivascular regions of chronically hypoxic mice and pulmonary hypertension (PH) being attenuated when homing of HSC to these regions is prevented.[6] The potential roles of mesenchymal stem cells (MSC), or mesenchymal progenitor cells (MPC), and endothelial progenitor cells (EPC) seems to be a paradox between pathogenic and therapeutic roles. There are many studies supporting both roles and all are likely to be correct reflections. The conditions in which these cells are recruited and the niche in which they reside are hugely influential of their characteristics. Furthermore, ex vivo manipulation of these cells may have significant effects on the properties of the cells.
The bone marrow is a niche where an expansive repertoire of stem and progenitor cells resides. In response to tissue injury or disease, these cells can be mobilized and are capable of homing to the lung. Such cells include HSC, MSC, and EPC and the key characteristics of each of these will be briefly described below with particular reference to their roles in pulmonary vascular disease. In addition, there are potentially many resident tissue progenitor cells that are either poorly characterized to date or have yet to be identified. A population of such cells has been identified in vascular walls and are, like most stem cell types, identified by their cell surface and intracellular marker expression, including CD133, CD44, and nestin. In the lung particularly these cells have been denoted side population cells (SP) and they can be further identified by their ability to efflux Hoechst 33342[7] due to a high expression level of the ATP binding cassette transporters (ABC) (e.g., ABCG2 enabling active efflux of the dye).[8]
HSC and the pulmonary circulation
Hematopoietic stem cells are perhaps the best characterized stem cells with their differentiation capacity fully delineated (Fig. 1). A single HSC is capable of differentiation to all blood cells which includes (1) myeloid cells encompassing monocytes, macrophages, neutrophils basophils, eosinophils, erythrocytes, megakaryocytes/platelets and dendritic cells, and (2) lymphoid cells comprising T-cells, B-cells, and natural killer cells. Mammalian hematopoiesis occurs in three distinct phases, the first two of which are primitive and definitive originate in the yolk sac where hemangioblasts develop. These multipotent precursors give ride to endothelial as well as primitive and definitive hematopoietic progeny. The emergence of the HSC is, however, uncertain and is postulated to be either from the yolk sac or the paraaortic splanchnopleure/aorta-gonad-mesonephros (P-Sp/AGM) prior to their detection in the fetal liver. For in-depth analysis of the current data for human HSC emergence, readers are encouraged to read the articles by Robertson et al., Dzierzak, Medvinsky et al., and Tavian et al.[9–12]
Adult HSC are round, nonadherent cells with a high nucleus-cytoplasm ratio and they reside primarily in the bone marrow and have the ability to leave the niche and home back to it. The stromal-derived factor-1 (SDF-1/CXCL12)/CXCR4 axis is critical for such homing and mobilization of HSC.[13] In the pulmonary circulation, this mechanism has also been shown to be important for homing of c-Kit+ hematopoietic progenitor cells to a perivascular niche in mice.[6] It is worth noting that in mice exposed to chronic hypoxia (CH) the expression levels of CXCR4, CXCR7, and CXCL12 are all elevated after onset of pulmonary hypertension. Administration of an antagonist of CXCR4 has been observed to prevent PH and reduce the associated vascular remodeling and perivascular accumulation of hematopoietic progenitor cells.[6] It will be interesting to see if similar mechanisms exist in humans.
Cell surface markers commonly used in combination to select for mononuclear HSC include CD34, CD133, and CD117 (c-Kit) in the human, in addition to a lack of differentiation markers CD2, CD3, CD14, CD16, CD19, CD24, CD56, CD66b, glycophorin A (Lin−). HSC can also be distinguished by their poor ability to accumulate metabolic fluorochromes such as DNA stain Hoechst 33342, rhodamine 123 or mRNA marker pyronin Y.[14,15] Low incorporation of mitochondrial dye (e.g., rhodamine 123) occurs due to its rapid efflux through activity of P-glycoprotein, a multidrug efflux pump.[16] Furthermore, the expression of integrin α6 (CD49f) in conjunction with Thy-1 (CD90), CD34 and the absence of CD45-RA and CD38 with low rhodamine 123 incorporation, has recently been shown to identify a single HSC capable long-term, multilineage reconstitution of an immunocompromised mouse though a single-cell intrafemoral transplant.[17]
Collection of HSC from blood samples requires a Ficoll-Paque density gradient centrifugation to deplete the erythrocytes and granulocytes from an anticoagulant-treated and diluted blood sample.[18] Ammonium chloride buffer can be used to lyse the erythrocytes in the sample enriching for HSC and other blood cells. Subsequent purification steps involve separation by cell surface marker expression using FACS or paramagnetic beads. Identification of HSC in tissue samples can be carried out by multicolor immunohistochemistry as detailed in this review.
Ficoll-Paque density gradient centrifugation[18]
It is imperative that all solutions and equipment must be sterile and used with proper aseptic technique.
Procedure
Place 15 ml of Ficoll-Paque solution into a 50 ml centrifuge tube
Carefully layer 30 ml of diluted blood on the Ficoll-Paque solution. Do not mix the blood and Ficoll-Paque solution
Centrifuge for 40 minutes at 400xg at 20°C
Collect the mononuclear cell fraction carefully using a Pasteur pipette at the interface between plasma and Ficoll-Paque and transfer into a clean centrifuge tube
If erythroid cells are present in the interface try treatment with 8% ammonium chloride or 3% diethylene glycol
Centrifuge cells for 10 minutes at 700xg
Add 5–20 ml of lysis solution to the pellet, mix the suspension, and incubate 5–10 minutes at room temperature
Centrifuge for 10 minutes at 700xg. Discard supernatant and proceed
Add 40 ml PBS/EDTA to wash the mononuclear fraction and centrifuge for 10 minutes at 300xg at 20°C
Discard the supernatant and repeat the wash with 40 ml PBS/EDTA and centrifuge again
Discard the supernatant and resuspend the mononuclear cells in 5–10 ml of PBS/0.5% BSA/2 mM EDTA and count the cells.
Functional activity of true HSC can be confirmed by in vitro differentiation to both myeloid and lymphoid lineages or be transplanted into immunocompromised mice and the long-term engraftment potential assessed. For more detail on intrafemoral injections for the transplantation of human HSC into immunocompromised mice please refer to the papers by Mazurier et al.[19] and McDermott et al.[20] Myeloid differentiation can be assessed by a methylcellulose colony forming unit assay. Methylcellulose is a semisolid media complete with cytokines supporting differentiation to myeloid cells (Stem Cell Technologies). Hematopoietic colonies grow in a three-dimensional nature and can be scored dependent upon the cell type they are formed from. A true HSC will be able to generate all myeloid cells from a single cell (thus a single myeloid colony forming unit containing granulocytes, erythrocytes, monocytes, megakaryocytes (CFU-GEMM)).
Methylcellulose assay for myeloid colony forming units
Procedure
After magnetic or FACS sorting carefully mix approximately 1×105 CD34+ cells in 2 ml of MethoCult GF H4434 (Stem Cell Technologies: 1% methylcellulose, 30% FBS, 1% BSA, 0.1 mM 2-mercaptoethanol, 2 mMl-glutamine, 50 ng/ml rhSCF, 10 ng/ml rhGMCSF, 10 ng/ml rhIL-3, and 3 U/ml rhEPO. Ensure that no bubbles are generated
Dispense the mix carefully into petri dishes using a syringe and blunt end needle and incubate in a humidified incubator at 37°C, 5 % CO2
Hematopoietic colonies can be enumerated and identified at days 14–21.
MSC and the pulmonary circulation
Mesenchymal stem cells are also referred to as multipotent mesenchymal stromal cells or multipotent progenitor cells (MPC) and are known to reside in niches where a turnover of mesenchymal-derived tissues occurs; this includes but may not be limited to the bone marrow, muscle, fat, skin, and cartilage. These cells demonstrate a great plasticity and, in the right conditions/niche, they are capable of changing from one lineage to another thus making characterization of this cell type particularly difficult. Due to the difficulties in defining MSC, the International Society for Cellular Therapy set a minimal criterion for putative MSC. To fulfill this criterion MSC must be adherent to plastic, they must express cell surface markers CD105, CD73, and CD90 and lack the expression of CD45, CD34, CD14 or CD11b, CD79α or CD19 and HLA-DR, and finally they should have the ability to differentiate osteoblasts, adipocytes, and chondroblasts in vitro.[21] Figure 2 shows a clear representation of MSC self-renewal and differentiation to all potential progeny.
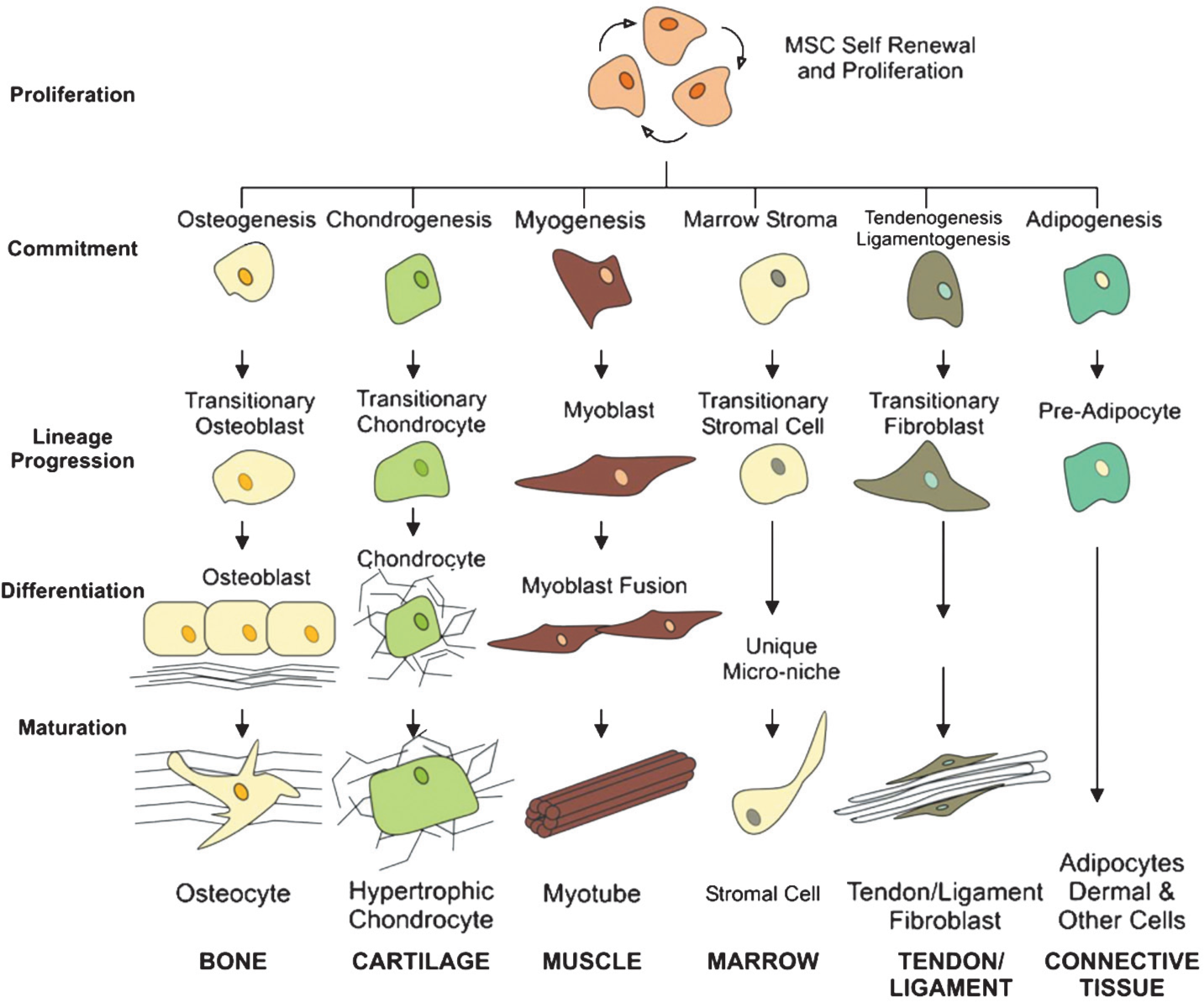
The mesengenic process. MSC self-renewal, proliferation, and potential lineage specific differentiation pathways are depicted in this diagram. MSC differentiate by committing, differentiating, and maturing in a lineage specific fashion. (Reproduced with permission from reference 84.).
Due to their great plasticity and homing capabilities, MSC have a huge potential as a therapeutic approach.[22] On the other hand, these same properties make them candidates for contributing to the vascular remodeling characteristic of PH. The therapeutic potential of MSC has been widely studied in the cardiovascular system where they are used as autologous cell therapy.[23] Recently an intravenous injection of MSC was used to treat experimentally induced PH in rats (monocrotaline model); significant improvements were observed in the right ventricular (RV) impairments in these rats. MSC were still alive and capable of endothelial cell differentiation in these rats 2 weeks post-transplantation.[24,25] The significant improvements in pulmonary arteriolar thickness are clearly seen in Figure 3.
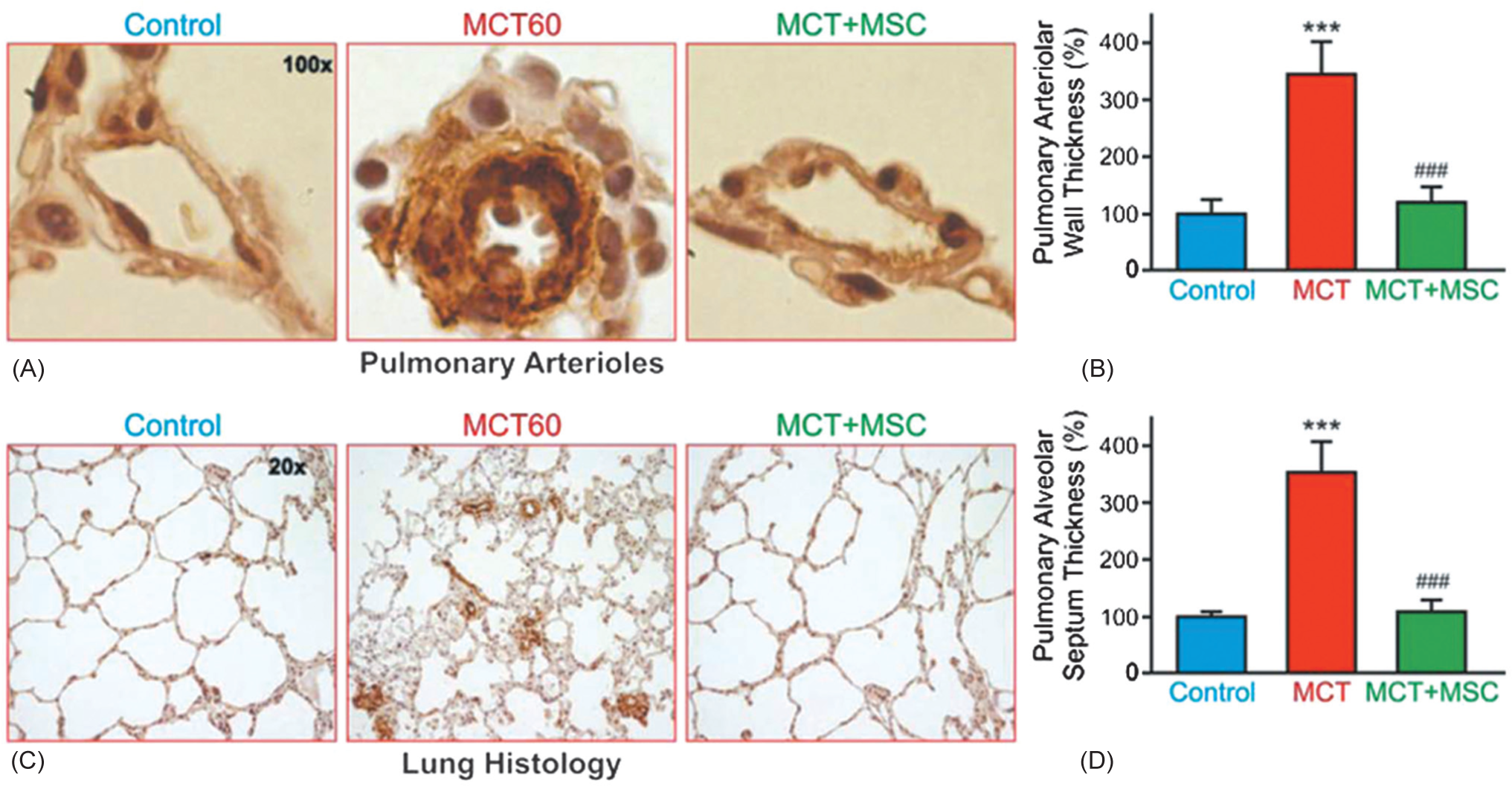
Immunoperoxidase images of paraffin sections of lung tissue stained with anti-α-smooth muscle actin antibody. (A) Representative pulmonary arterioles of control, monocrotaline treated (MCT60), and monocrotaline and MSC treated (MCT + MSC) rats. Images were acquired using a x100 objective. (B) Pulmonary arteriolar wall thickness (in %) of control, MCT60, and MCT + MSC rats. Values are means ± SD. ***P<0.001 vs. the control group; ###P<0.001 vs. the MCT60 group. (C) Immunoperoxidase images of paraffin sections of lung tissue stained with anti-α-smooth muscle actin antibody. The lung histology of rats in the control, MCT60, and MCT + MSC is shown. Images were acquired using a x20 objective. (D) Pulmonary alveolar septum thickness (in %) of control, MCT60, and MCT+MSC rats. Values are means ± SD. ***P<0.001 vs. the control group; ###P<0.001 vs. the MCT60 group. (Reproduced with permission from reference 25).
MSC also have the potential to be a vehicle for gene therapy for lung disease due to their preferential homing to the lung. Several studies have now investigated the use of MSC as a tool for drug/gene delivery and considerable improvements in the pathogenesis of PH have been observed. This approach has been used to deliver agents including angiopoietin-1 for acute lung injury,[26] endothelial nitric oxide synthase (eNOS) for PAH-related RV impairment,[27] heme-oxygenase-1 for PH[28] calcetonin gene-related peptide in vascular smooth cell proliferation,[29] and prostacyclin-synthase for PH.[30] They have been similarly exploited in other diseases with positive benefits observed, for example hetatocellular carcinoma[31] and metastatic cancers.[32]
There are now several studies demonstrating a contribution of MSC to the pathogenesis of PH. The vascular adventitia itself is known to contain MSC/MPC[33] and the vasculature is also known to contain a side population of CD45−, c-kit−, CD11b−, CD34−, CD14−, CD44+, CD90+, CD105+, CD106+, CD73+, and Sca-I+ with adipogenic, osteogenic, and chrondrogenic potential.[34] The exact roles of such resident stem cells are yet to be fully elucidated and it is established that the environmental niche is critical in regulating the maintenance and differentiation of stem cells, thus making the pathogenic roles of such cells difficult to fully understand in animal models and in vitro conditions. Fibrocytes are a progenitor cell derivative of an MSC capable of differentiating into fibroblasts and myofibroblasts. Circulating fibrocytes have been shown to contribute to the deposition of extracellular matrix in pulmonary fibrosis.[35] Fibrocytes and MSC have also been shown to be recruited and contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension.[36,37] Inhibition of CXCR4 signaling is a potential therapeutic approach in hypoxic induced PH as evidence suggests that its inhibition prevents the mobilization of bone-marrow-derived MSC to the pulmonary vasculature.[38] Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELM α) may also act as a chemotactic agent for bone-marrow-derived MSC-mediated remodeling of the pulmonary vasculature in chronic hypoxia-mediated PH.[39]
Pulmonary hypertension is known to be an extremely complex phenomena with multiple pathways existing and contributing to the various aspects of the disease.[40–44] While it may be impossible to define the triggering event, research continues to show interaction of the pathways. Recently the roles of seretonin signaling and MPC were linked, with the expression of 5-HT2B receptors on bone-marrow-derived MPC shown to be critical for the development of PAH in mice.[45] Furthermore, mesenchymal cells with all the traits of an MSC have been found to have a high presence in endarterectomized tissues from patients with chronic thromboembolic pulmonary hypertension (CTEPH).[46] The role of fibrocytes, a mesenchymal-derived progenitor cell, in the pulmonary vasculature is comprehensively reviewed by Stenmark et al.[47]
Functional activity of putative MSC can be confirmed by verification of their differentiation capacity once their cell surface markers expression has been assessed. A true MSC should be capable of differentiation to adipocytes, chondrocytes, osteocytes, and myocytes. Complete kits designed for adiopcyte, chondrocyte, and osteocyte differentiation from MSC are commercially available or medias can be made in-house. The protocols described below are adapted from the commercially available Invitrogen protocols and the paper by Reger et al.[48] Figure 4 demonstrates a basic characterization of bone-marrow-derived MSC by FACS and their differentiation to adipocytes and osteocytes. In addition, human MSC have recently been shown to be an excellent source of SMC for arterial engineering.[49,50] The differentiation is pushed by the addition of transforming growth factor β (TGFβ) and cells acquire a contractile smooth muscle cell phenotype.[51] With the ever increasing need for rapid tests to confirm differentiation to validate the stem cell phenotype, Boucher et al. developed a PCR screen designed to detect the early stages of mesenchymal stem cell differentiation.[52]
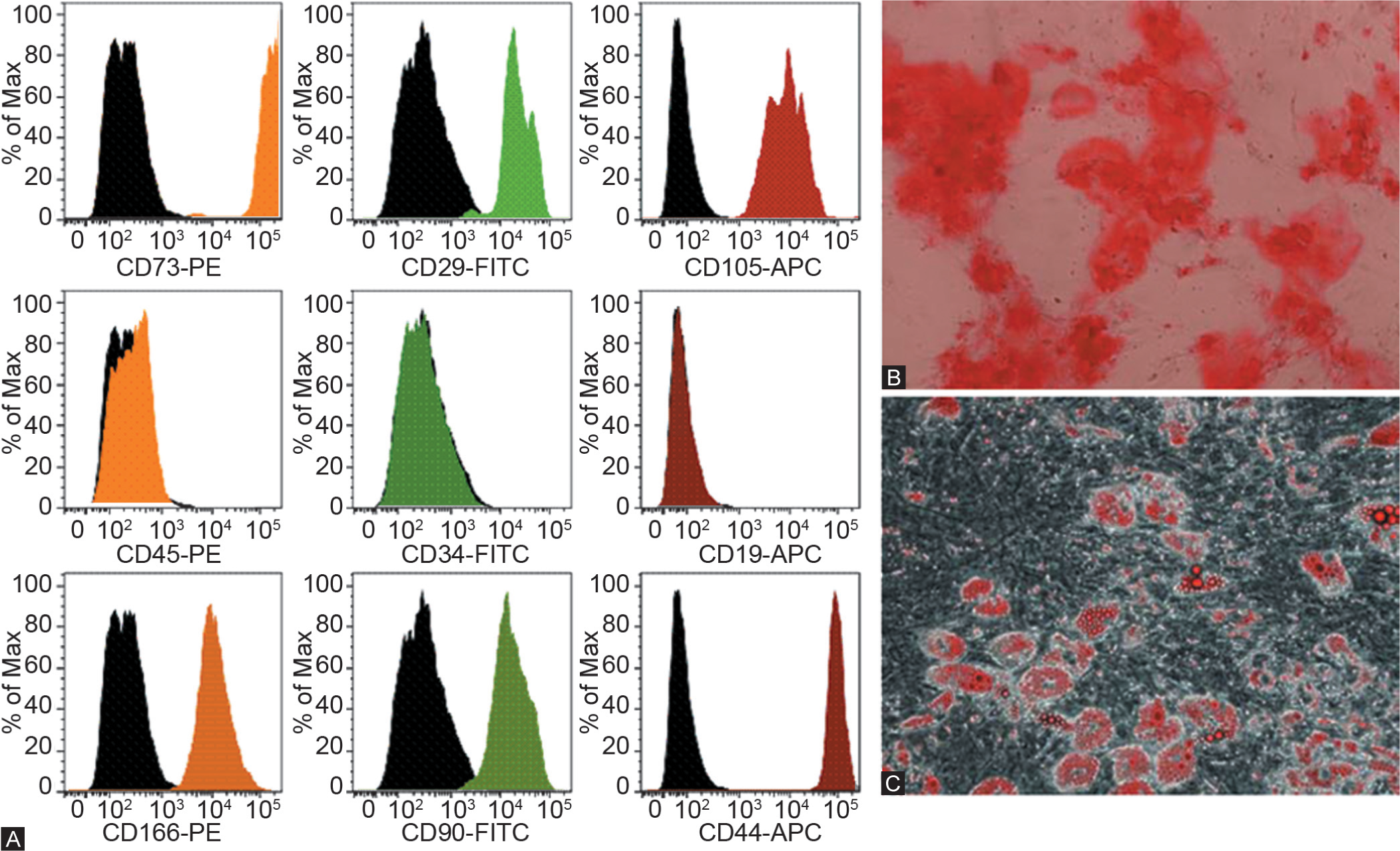
Characterization of bone-marrow-derived MSC. This figure highlights a core set of marker expression patterns used in flow cytometry to isolate putative MSC (A) and differentiation to osteocytes (B) stained with alizarin red and adipocytes (C) stained with oil red O.
Adipocyte differentiation (adapted from reference 48)
Grow putative MSC in a suitable growth medium (e.g., αMEM minus deoxy- and riboxynucleosides, 15% FBS, 1% Pen/Strep, 1% Glutamax) to 60–80% confluence. Aspirate medium and gently wash the cells in PBS.
Procedure
Add 2–7 ml of prewarmed TrypLE™ Express (Invitrogen), sufficient to cover the culture surface and incubate until cells have detached (~3–8 minutes at 37°C)
Gently triturate the detached cells to form a single cell solution using a wide bore glass pipette
Pellet the cells at 100xg for 5 minutes
Cell viability and total cell density may be determined at this stage using Trypan Blue Stain and counting the cells using a hemocytometer (or other suitable cell counting method)
Resuspend the pellet in appropriate volume of prewarmed MSC Media and seed the putative MSC into the selected culture vessel at a density of 1×104 cells/cm2. Culture vessels should be selected as follows:
For classical stain differentiation assay use a 12-well plate
For gene expression profiling use a T-75 flask
For immunocytochemistry use a 16-well Culture Well chambered cover glass
Incubate in a humidified atmosphere at 37°C, 5% CO2 for 2 hours to 4 days
Replace media with prewarmed adipogenesis differentiation medium consisting of αMEM minus de- and rib-oxynucleosides, 15% FBS, 1% Pen/Strep, 1% Glutamax, 0.5 μM dexamethasone, 0.5 μM isobutylmethylxanthine and 50 μM indomethacin and continue incubation changing media every 3–4 days. It should be noted that MSC may continue to undergo limited expansion as they differentiate
After determined periods of incubation (up to 21 days) staining the cultures with oil red O will confirm the presence of adipocytes.
Oil red O stain analysis
Procedure
Prepare the oil red O stain as follows: 0.5% oil red O stock solution: 2.5 g oil red O into 500 ml isopropyl alcohol and dissolve completely. From the stock make a working solution of three parts 0.5% oil-red-O Stock; two parts PBS. Mix this thoroughly and wait 10 minutes. Filter and wait a further 10 minutes before use
After 21 days of differentiation aspirate the medial and wash twice with PBS
Add 2 ml of neutral buffered formalin (NBF) and incubate for 1 hour at room temperature
Aspirate the NBF and wash the wells with 2 ml PBS.
Add 2 ml of oil red O and incubate for 20 minutes at room temperature, then aspirate
Rinse the wells twice with 2 ml of PBS and aspirate
Add a final 2 ml of PBS and examine plate on an inverted microscope for evidence of fat deposits and/or bone differentiation.
Chondrocyte differentiation (adapted from reference 48)
Procedure
Repeat steps 1–5 from the adipogenesis differentiation protocol
Resuspend the pellet in appropriate volume of prewarmed chondrocyte media with cytokines (DMEM High Glucose, 50 μg/ml L-ascorbic acid-2-phosphate, 40 μg/ml L-Proline, 100 μg/ml sodium pyruvate, ITS Culture Supplement (BD Biosciences) 10 ng/ml rhTGF-β3, 10 nM dexamethasone, 500 ng/ml rhBMP-2 or rhBMP-6) and generate a cell solution of viable cells at a density of 1.6×107 cells/ml
Count cells and assess viability. Adjust to approximately 400 viable cells/μl with chondrocyte media with cytokines
Transfer approximately 200,000 MSC in 500 μl to a 15 ml falcon tube and centrifuge at 450xg for 10 minutes
Do not resuspend or remove the media and incubate in a humidified atmosphere at 37°C, 5% CO2
Fresh medium should be added every 3 days
After determined periods of incubation the chondrogenic pellets can be confirmed by staining with alcian blue or safranin O (21 days).
Alcian blue stain analysis
Procedure
After differentiation for 21 days remove medium, wash cells and fix in 4% PFA for 10–30 minutes
Wash fixed cells and stain with 1% alcian blue solution prepared in 0.1 N HCL for 30 minutes
Wash x3 with 0.1 N HCl, add distilled water to neutralize the acidity and visualize under light microscope. Blue staining is indicative of proteoglycan synthesis by chondrocytes.
Osteocyte differentiation (adapted from reference 48)
Procedure
Repeat steps 1–8 from the adipogenesis differentiation protocol, except:
Seed the MSC into the selected culture vessel at a density of 5×103 cells/cm2
Replace media with prewarmed osteogenesis differentiation media (.αMEM minus deoxy- and riboxynucleosides, 15% FBS, 1% Pen/Strep, 1% glutamax, 10 nM dexamethasone, 20 mM β glycerol phosphate, 50 μM L-ascorbic acid-2-phosphate)
After specific periods of cultivation, osteogenic cultures can be processed for alkaline phosphatase staining (7–14 days) or alizarin red S staining (>21 days).
Alizarin red S stain analysis
Procedure
After 21 days, remove media and wash once in DPBS. Fix with 4% PFA for 10–30 minutes or NBF for 1 hour at room temperature
Wash fixed cells twice with distilled water and stain with 2% alizarin red S solution for 2–3 minutes. Alizarin red should be prepared as follows: 1 g alizarin red S in 100 ml DI water and the pH should be adjusted to 4.1 and 4.3 using 0.1% ammonium hydroxide before filtering
Wash x3 with distilled water and visualize under light microscope.
Myocyte differentiation (adapted from reference 49)
Procedure
Repeat steps 1–8 from the adipogenesis differentiation protocol, except:
Seed MSC into the selected culture vessel at a density of 2.4×103 cells/cm2
Replace media with prewarmed MesenPro RS™ with 1 ng/ml TGFβ and change media twice a week
After approximately 14 days cultures can be processed for expression of smooth muscle marker including, but not exclusively; SM-22α (transgelin), smooth muscle-myosin heavy chain (SM-MHC), smoothelin, and caldesmon.
Simplified PCR screen for early stages of mesenchymal stem cell differentiation (adapted from reference 52)
Procedure
Follow suitable differentiation protocols as outlined above
Harvest RNA from cultures after 7 days of differentiation.
Isolate total RNA and synthesize first strand cDNA using an appropriate commercially available kit
Run the PCR using the primer pairs listed below and run agarose gel to resolve the PCR products.
Primers required for screen
B2M (β-2-microglobin), a housekeeping gene, (314 bp) F‘: GCGTACTCCAAAGATTCAG, R‘: CAAACCTCCATGATGCTG: CD73 (5′ ecto nucleotidase) an MSC cell surface marker, (414 bp) F‘: CAATTGTCTATCTGGATGGC, R‘: GACACTTGGTGCAAAGAAC: RGC32 (response gene to complement 32) an early osteocyte cell marker, (166 bp) F‘: GCCACTTCCACTACGAGGAG, R‘: GCTGGGGTAGAGTCTGTTGG: FABP4 (fatty acid-binding protein 4) an early adipocyte cell marker, (215 bp) F‘: TCATACTGGGCCAGGAAT, R‘: TCCCTTGGCTTATGCTCT: SPP1 (bone sialoprotein 1) an early chondrocyte cell marker, (229 bp) F‘: CTCCATTGACTCGAACGACTC R‘: CAGGTCTGCGAAACTTCTTAGAT.
EPC and the pulmonary circulation
EPC exist in a hierarchy with individual subdivisions identified by the ability of the cell to divide in a clonogenic nature and to proliferate.[53,54] EPC is the all-encompassing term used to refer to the entire group of these cells but really should be restricted cells with the correct cell surface marker expression and with the ability of form de novo vessels. The first recognition of EPCs was back in 1997 when a population of circulating CD34 positive cells capable of in vitro differentiation and de novo vessel formation was identified.[55] Prior to this discovery, new blood vessel formation was thought to rise from the proliferation, migration and remodeling of mature endothelial cells. EPC function in the pulmonary vascular system is, however, currently controversial. The diagram in Figure 5 overviews this current paradox.
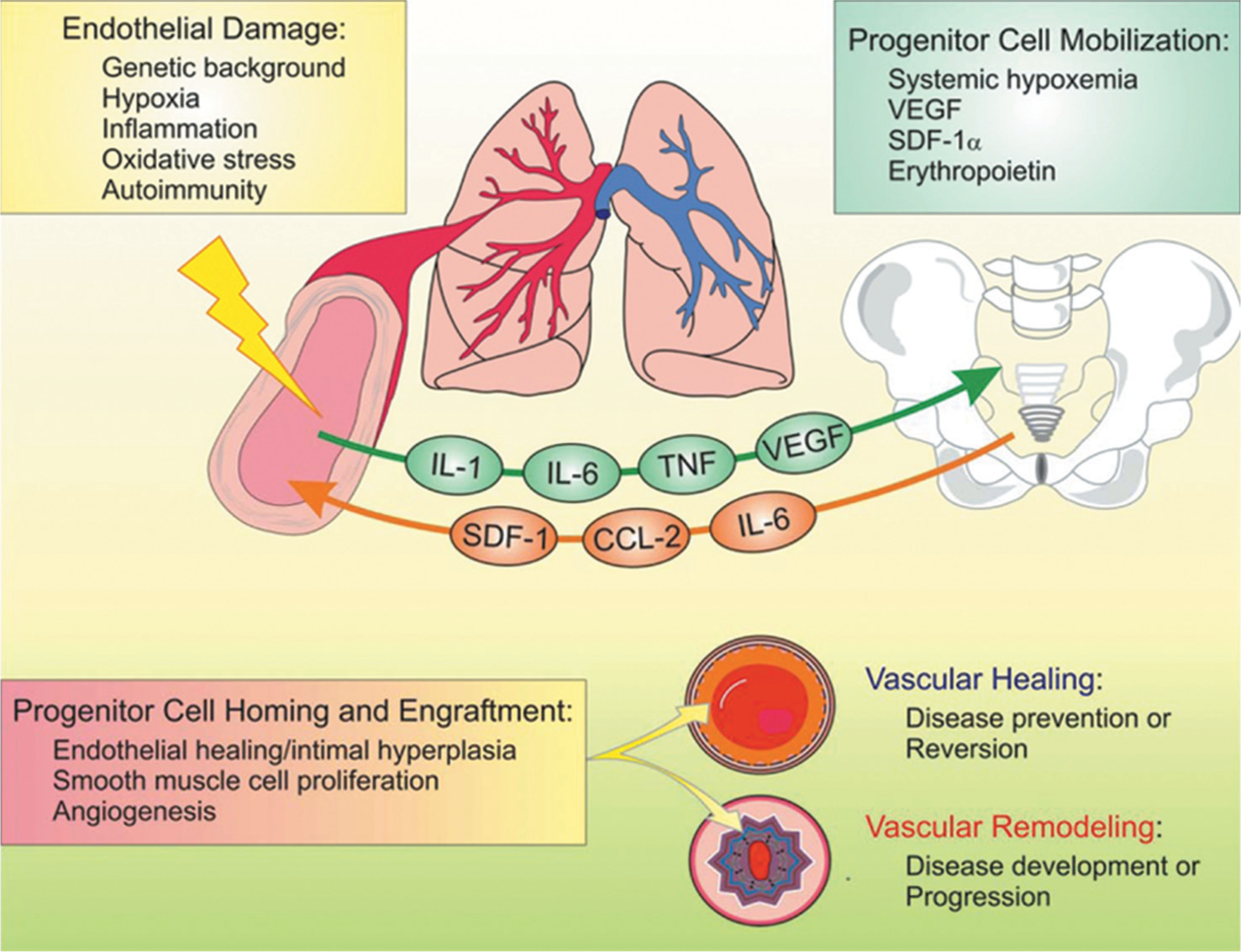
The EPC paradox: Contribution to disease development or vascular healing in pulmonary hypertension. IL: Interleukin; TNF: Tumor necrosis factor; VEGF: Vascular endothelial growth factor; SDF: Stromal-cell-derived factor; CCL: Chemokine (C-C motif) ligand.
In a monocrotaline (MCT)-induced canine model of PH, neovascularization and a reduction in mean pulmonary arterial pressure (mPAP), cardiac output (CO), and pulmonary vascular resistance (PVR) were observed after transplantation of ex vivo expanded autologous EPC from peripheral blood.[56] Similar results where EPC engraft, restoring microvasculature structure, and function were observed in MCT induced PH in rats.[57] In mice, the endogenous erythropoietin/erythropoietin receptor (Epo/EpoR) system is important in recruiting EPC to the pulmonary vasculature and a therapeutic benefit is observed with an attenuation of the development of PH.[58] In support of a therapeutic benefit of EPC it has been noted that a severe depletion of circulating EPCs correlates to the development of chronic lung disease, idiopathic pulmonary fibrosis (IPF) and PH.[59,60] Furthermore, in IPF patients who developed secondary PH, the depletion of EPC was comparatively worse implicating a clinical benefit of therapies positively modulating EPCs.[60] In 2007, the therapeutic benefit of EPC in PH was explored further by the initiation of clinical trials. A prospective, randomized trial comparing the effects of conventional therapy with or without the intravenous infusion of EPC in patients with IPAH demonstrated significant improvements in the mean walk test, mPAP, PVR, and CO in the patients with the EPC treatment.[61] There is also evidence suggesting that the clinical benefit of prostanoids may be due to/enhanced by EPC.[62] With evidence supporting the number of circulating EPC correlating to cardiovascular risk, a group designed a disposable microfluidic platform capable of selectively capturing and enumerating EPC directly from human whole blood. Using this chip they confirmed a 50% reduction in EPC in PAH subjects versus matched controls.[63] This EPC capture chip may be used in the screening and monitoring of patients with PAH in the future. EPC are capable of being mobilized in response to vascular injury. For example, VEGF is known to effectively mobilize EPC and potently induces angiogenesis; shear stress can also promote EPC differentiation into mature endothelial cells.[64] Homing of EPC to a site of injury is likely due to cell surface expression of chemokine receptor CXCR4 and the chemoattractant pull of SDF-1, released from EPC and platelets. Furthermore, high levels of β2 integrins on EPC can interact with their ligands P-selectin, E-selectin, and ICAM-1 that are expressed on EPC.[65]
Despite the wealth of research supporting a therapeutic benefit of EPC, there are also studies providing evidence for a pathogenic role of these cells. The contribution of progenitor cells to pulmonary vascular remodeling was recently reviewed and readers are encouraged to read Yeager et al. for a detailed discussion.[42] Briefly, EPCs have been found to contribute substantially to the development of plexiform lesions in PH,[66] endothelial to mesenchymal transition resulting in fibrosis,[67,68] and to the fibrotic embolism in patients with CTEPH.[69] The increased expression of CXCR4 and SDF-1 in plexiform lesions from patients with idiopathic pulmonary hypertension is nicely demonstrated in Figures 6 and 7, clearly showing the characteristics of CD133, von Willebrand factor, CCD34 and CD146 positive late outgrowth EPC isolated from the plexiform lesions.[66]
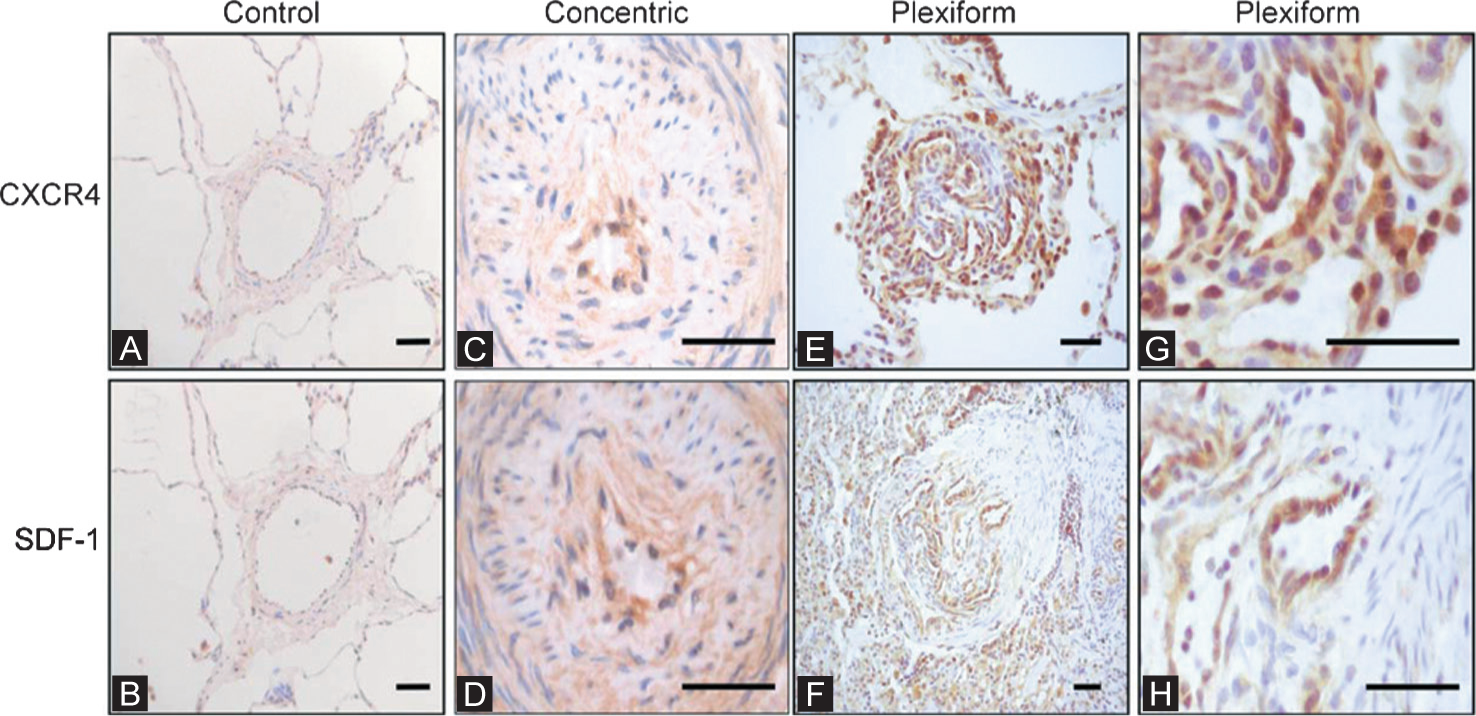
Representative photomicrographs of peripheral lung tissue from (A and B) normal control lung and (C–H) a patient with pulmonary arterial hypertension (PAH); samples were immunostained for CXCR4 and stromal-cell-derived factor (SDF)-1. Minimal staining is seen in normal lung. (C and D) In PAH lung low-level staining was observed in concentric intimal lesions. (E and G) CXCR4 expression was generally increased in the lung parenchyma of patients with PAH, but was also present in the endothelium of plexiform lesions. (F and H) SDF-1 was less prevalent but showed clear staining of the endothelium of plexiform lesions. Scale bars: 50 μm. (Reproduced with permission from reference 66).
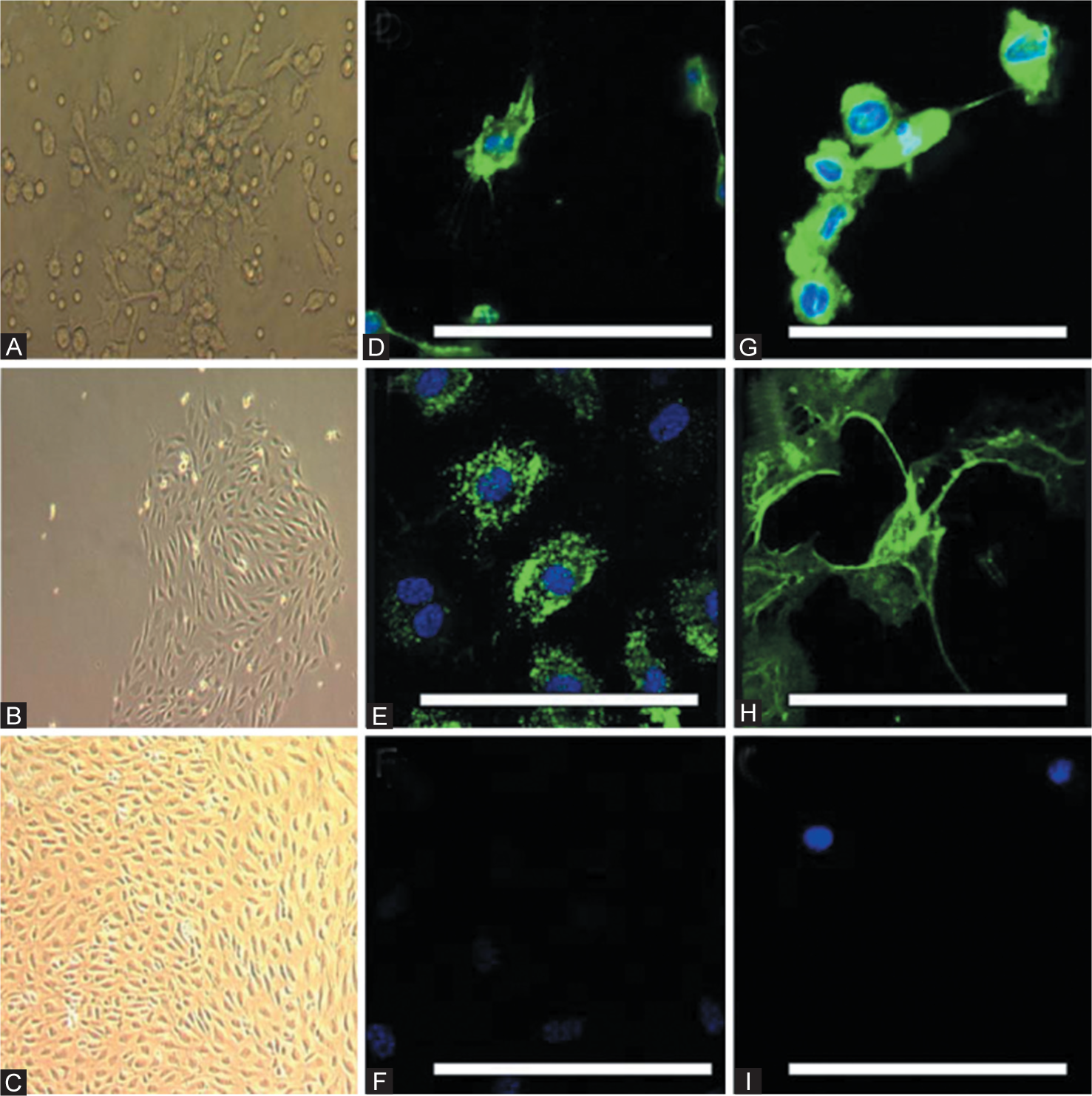
Phase-contrast photomicrographs of cultured late-outgrowth endothelial progenitor cells (EPCs) showing (A) a colony-forming unit at 3 days, and a late-outgrowth colony at (B) 2 weeks and (C) 3 weeks. Confocal immunofluorescence images using conjugated fluorescein isothiocyanate (green) demonstrate that occasional cells were positive for (D) CD133 and that the majority of cells were positive for (E) von Willebrand factor, (G) CD34, and (H) CD146. Nuclear counterstaining was performed with 4′,6-diamidino-2-phenylindole (blue). (F and I) Isotype controls for anti-mouse and anti-rabbit secondary antibodies. Scale bars: 50 μm. (Reproduced with permission from reference 66).
Mead et al.[70] describe in detail the isolation and characterization of EPC. Table 1 provides a detailed comparison of the cellular markers and vasculogenic activity of derivatives of EPCs. Identification of functional EPC can be carried out using acylated-LDL (low-density lipoprotein), readily uptaken by endothelial cells through the “scavenger cell pathway” of LDL metabolism.[71] By examining the fluorescent signals, uptake of DiI-Ac-LDL (1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate) has been used to demonstrate how putative progenitor populations take on properties of functional endothelial cells.[72,73] EPC functionality may also be assessed through the formation of bona fide tubes in vitro in Matrigel; this characteristic is unique to endothelial cells.[74] The most rigorous test of putative EPC is the engraftment into, or de novo formation of, functional blood vessels in vivo. An intravenous injection of EPCs directly into injured tissues, or implanted within a matrix or tumor environment, is utilized, and by prelabeling the cells of interest with a fluorescent dye[75] or transducing cells with a viral-driven fluorescent reporter,[76] precise microscopic examination of vasculature within these injured or implantation sites can be carried out via confocal image analysis. Transmission electron microscopy may be used to further demonstrate the ultrastructure of the tube formed in the in vitro assay and the blood vessels formed in the in vivo assay. Examples of both in vitro and in vivo vessel formation by EPC are shown in Figure 8.
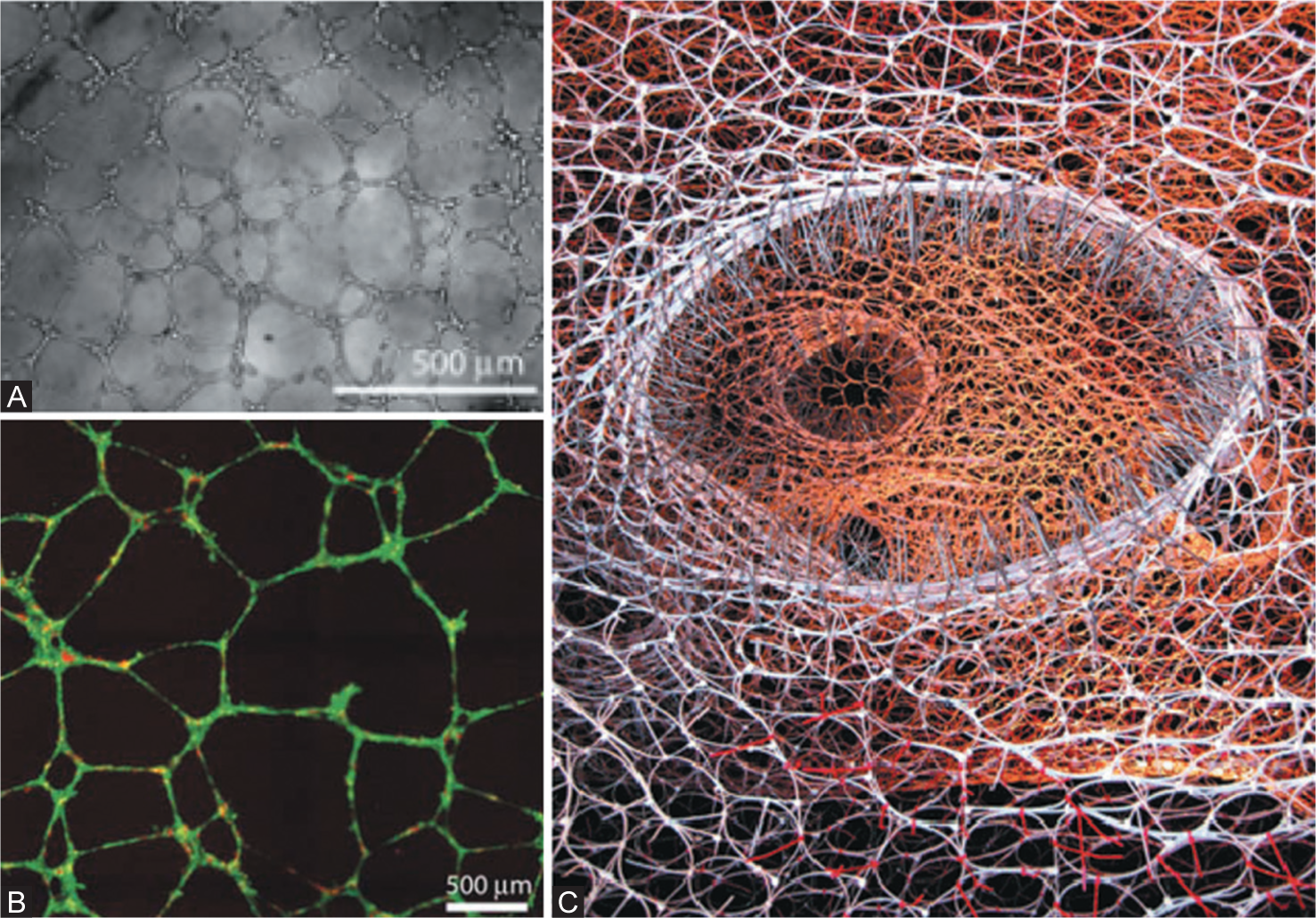
EPC tube formation in vitro. Bright-field image of human microvascular endothelial cells (HMECS) forming vascular networks on Matrigel after 12 hours (A). Fluorescent images of CellTracker Red-labeled HMECs at 9 hours on Matrigel (B). A sculpture designed from five snapshots of a computer simulation of branching morphogenesis illustrating the forces lung cells exert as they form capillaries. (A and B are reproduced with permission from reference 88, C is reproduced with permission from reference 89).
Endothelial progenitor cells populations
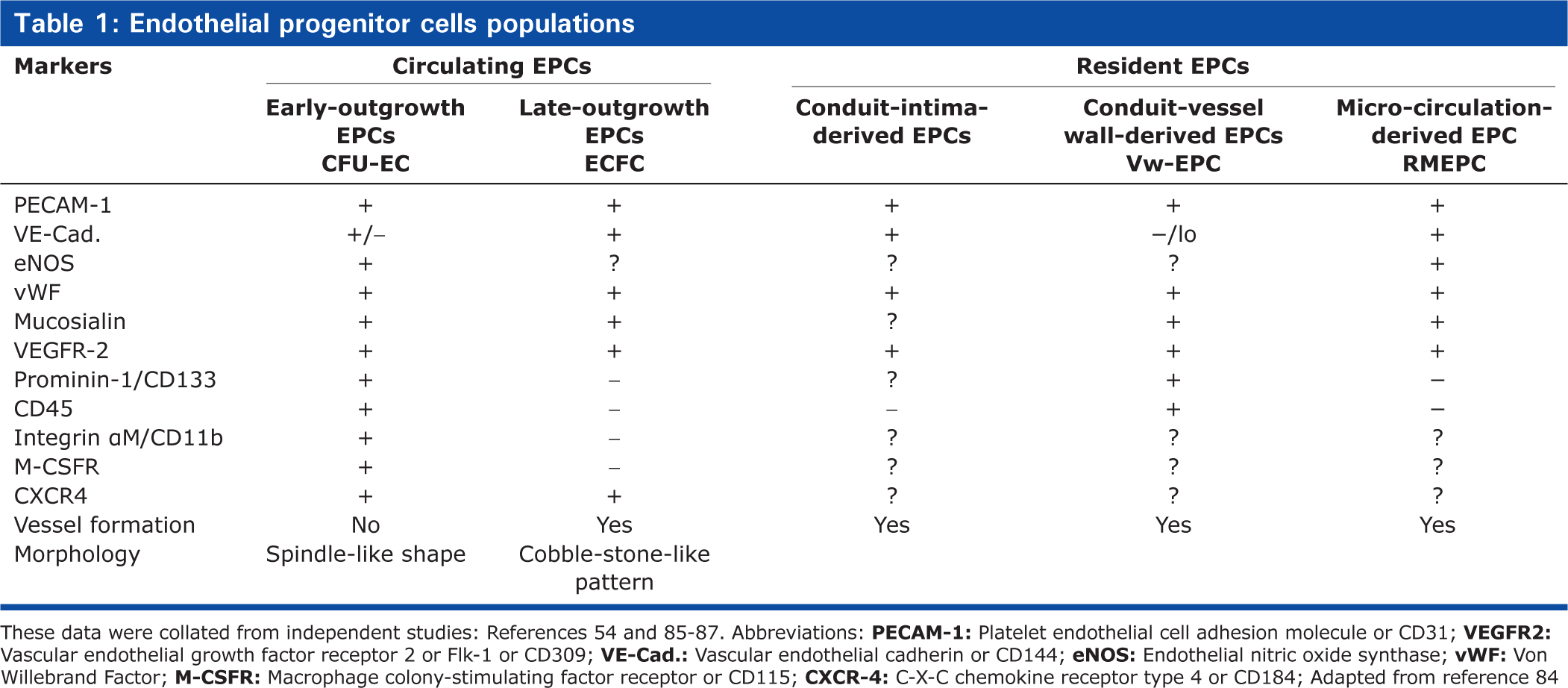
These data were collated from independent studies: References 54 and 85–87. Abbreviations:
Uptake of DiI-Ac-LDL (adapted from reference 53)
Procedure
Propagate endothelial cells on glass-bottom chamber slides
Aspirate culture medium and wash the cells with PBS
Incubate the attached cells with 10 mg/ml DiI-Ac-LDL in complete endothelial growth media (e.g., EGM, Lonza) for 4 hours at 37°C
Aspirate media and wash twice in PBS to remove free DiI-Ac-LDL
Fix the cells with 3% PFA/PBS for 10 minutes
Mount the slide with Vectashield with DAPI (Vector Lab)
Examine the slide with rhodamine filter for DiI-Ac-LDL and Hoechst filter for DAPI in a fluorescence microscope and acquire images.
Assessment of tube formation in vitro (adapted from reference 54)
Procedure
Seed putative progenitor cells at 2×104 cells/cm2 in Matrigel (BD Biosciences)-coated (100 μl/cm2) 48-well plates, ~20,000 cells/well. Each well should contain 400 μl standard culture medium. Lung fibroblasts are suitable to use as a control. Each putative progenitor cell phenotype should be seeded triplicate
Incubate the plates in a humidified atmosphere at 37°C with 5% CO2-21% O2. Media should be changed 4 days after initial seeding
Take images on phase-contrast microscope at x10 magnification at 8, 24, and 48 hours, and at 1 week after seeding
Determine the network formation by counting the number of branches that connect 2 distant cells
Average counts from three wells at x10 magnification were classed as one experiment; three separate experiments should be completed to determine a result.
Assessment of de novo vessel formation in vivo (adapted from reference 54)
Procedure
Resuspend the cells in 1.7 ml microtubes containing 250 μl standard culture medium at 4°C. The cell density should be 375,000 cells per tube
Mix the cell-containing solution with 500 μl of unpolymerized Matrigel at 4°C. The cold temperature is critical prevent polymerization of the Matrigel
Mildly sedate a rat (e.g., i.p. ketamine 75 mg/kg)
Inject the 750 μl of the cell/Matrigel mix subcutaneously into left and right lumbar abdominal regions of rats using a 23-gauge needle. This will create two plugs per animal. The injected mixture polymerizes at body temperature and forms a plug following subcutaneous contact
Controls should be carried out in parallel consisting of a Matrigel mix with no cells
Anesthetize the animal using sodium pentobarbital (i.p. 50 mg/kg)
Excise the Matrigel plugs from the abdominal wall of animals at 4 and 10 days postinjection and fix by immersion in 4% PFA for 18–24 hours
Dehydrate the fixed plugs in ethanol and embed in paraffin
Attach the tissue block to a plastic block by melting the back of the tissue block with a warm spatula and firmly pressing the two together and then use a standard microtome to cut 5 μm sections
Stain the sections with hematoxylin and eosin (H and E) following a standard protocol
Examine the stained sections with a light microscope to count the total number of tubes containing red blood cells (i.e., blood vessels) within the gel.
General techniques for the identification of adult stem and progenitor cells in the pulmonary vasculature
The identification of adult stem and progenitor cells require a rigorous characterization process, especially as there is no single feature or marker specific to each stem cell type capable of identification alone. Identification and isolation of cells based upon a panel of cell surface and cytosolic markers can be followed by functional assays to confirm the self-renewal and differentiation potential of the isolated cell population. Table 2 summarizes the cell surface and cytosolic markers known to be present (or absent) on the stem and progenitor cells. The expression of these markers can be utilized by a variety of techniques to identify and isolate stem cells in the pulmonary vasculature including immunohistochemistry, immunofluorescence, reverse transcription polymerase chain reaction (RT-PCR), protein detection by Western blot and flow activated cell sorting (FACS). Another characteristic of stem cells is their telomerase activity. Telomere length or telomerase activity measurements serve as a criterion to identify stem and progenitor cells. Telomeres are portions of genetic material involved in stabilizing the chromosome ends.[77,78] Long telomeres are prominently found in rapid-growing cells while short telomeres are associated with replicative senescence and loss of stem cell proliferation capacity in vitro. Finally, the clonogenic potential of a cell is a rigorous test indicative of their stem/progenitor capacity, a single stem cell having the ability to divide and form a colony of cells in the absence of other cells.[53,79] A clonogenic assay has been utilized effectively to delineate a hierarchy of EPCs.[80]
Cell surface markers expression of stem and progenitor cells known to be present in the pulmonary vasculature

Immunhistochemistry
Immunhistochemistry (IHC) is used to detect selected antigens within a tissue section by use of specific antibodies raised against the antigen in question. The technique can be used to investigate the distribution and localization of stem cells in tissue sections from the lung and pulmonary vasculature. There are two steps to the process: (1) preservation of the tissue; (2) detection of antigens specific to stem and progenitor cells.
Frozen section preparation
Procedure
Lung tissue should be carefully harvested and washed in chilled PBS. Place the sample in 30% sucrose/PBS solution at 4°C for 8–10 hours, then wash twice in PBS (5 minutes each)
Make sure all samples, bags and cryomolds are accurately labeled
To freeze the sample prepare a mix of dry ice and 2-methlylbutane in a plastic beaker, the temperature should approximately −40°C
Blot the excess sucrose from the tissue with gauze
Fill a cryomold halfway with Optimal Cutting Temperature (OCT) compound (Sakura Inc.) ensuring there are no bubbles in the OCT
Place the tissue in the OCT with the desired area of analysis facedown. Continue to fill the remainder of the mold with OCT
Carefully immerse the entire mold into the 2-methylbutane for 30–40 seconds, remove, place in a labeled plastic bag and store at −80°C
Prior to sectioning, the samples should be placed in a cryostat for 1 hour to bring them to 20°C. Cut the tissue block to 5–8 μm thickness and place the sections on microscope slides. Store the slides at −20°C until use.
Immunohistochemistry—detection
Procedure
Thaw the frozen tissue sections from −20°C freezer at room temperature
Fix the tissue sections in 4% PFA/PBS for 5–10 minutes and then wash with PBS for 3×5 minutes
Slides may be incubated for 5–10 minutes in 0.1–1% hydrogen peroxide diluted in PBS, deionized H2O or methanol to quench endogenous peroxidase activity. Wash twice in PBS
Incubate sections for 1 hour in 1.5% normal blocking serum in PBS + 2% serum + 0.1% Triton X-100 (for permeabilization). The blocking serum should be derived from the same species as the secondary antibody was raised
Incubate with primary antibody for 1 hour at room temperature or overnight at 4°C. Optimal antibody concentration should be determined by titration; recommended range is 0.5–5.0 μg/ml diluted in PBS with 1.5% normal blocking serum. Wash with PBS for 3×5 minutes
Immunoperoxidase staining using the ABC staining system (Santa Cruz Biotechnology). Incubate for 30 minutes with biotin-conjugated secondary antibody as provided, or at approximately 1 μg/ml diluted in PBS with 1.5% normal blocking serum. Wash with PBS for 3×5 minutes
Incubate for 30 minutes with avidin biotin enzyme reagent. Wash with PBS for 3×5 minutes
Incubate in peroxidase substrate, as provided, for 0.5–10 minutes, or until desired stain intensity develops. Individual slides should be monitored to determine the best development time. Wash sections in deionized H2O for 5 minutes. If desired, counter-stain in Gills formulation #2 hematoxylin for 5–10 seconds. Immediately wash with several changes of deionized H2O
Dehydrate through alcohols and xylenes as follows: Soak in 95% ethanol 2 × 10 seconds, then 100% ethanol 2×10 seconds, then xylene 3×10 seconds. Wipe off excess xylene and immediately add 1–2 drops of permanent mounting medium (e.g., CC/Mount, Sigma), cover with a glass cover slip and observe by light microscopy
Fluorescent-activated cell sorting
Fluorescent-activated cell sorting (FACS) using antibodies for specific protein markers can be used to serrate cells one at a time by the light scatter of fluorescent labeled antibodies/probes. Advancements in FACS equipment have enabled multicolour detection enabling multiple fluorescent probes to be detected on a single cell. Cells can be sorted in sterile conditions ready for further analysis. Magnetic bead separation can also be used and readers are encouraged to read Wills et al. for more detail.[81]
Procedure
Create a single cell suspension by using Accutase (Innovative Cell Technologies) or TrypLE (Invitrogen) or other suitable agent. Accutase helps to preserve cell surface antigens where trypsin-based products may have better dissociation activity
Wash cells once or twice with 2 ml cold PBS by centrifuging at 150–300xg at 4°C
Resuspend cells in Buffer (PBS + 1% BSA + 0.1% NaN3) and aliquot into 1-100×105 cells per 100 μl (total volume once antibodies added) in Falcon #2052 or #2054 tubes on ice
Add 20 μl of monoclonal antibody (1–10 μg/ml final concentration) or isotype control antibody. Antibodies will need to be titrated to determine optimal concentration
Incubate 30 minutes on ice or at 4°C, in the dark if using directly conjugated antibodies
Wash with 2 ml cold azide buffer
Resuspend in 100 μl of secondary antibody (e.g., fluorophore conjugated goat antimouse IgG). Skip to step 10 if using directly conjugated mAb
Incubate 30 minutes on ice in the dark and then wash with azide buffer
Resuspend in 100–500 μl cold wash buffer and add equal volume of cold buffered 3% PFA to fix cells and analyze or; leave in wash buffer and analyze live, add propidium iodide, 0.5 μg/ml final concentration. Live cells can be sorted by a Becton Dickinson FACS Aria machine into tubes containing 100% serum. Ideal final concentration of cells should be 1×106/ml.
Immunofluorescence imaging
Immunofluorescent labeling is essentially a combination of the three approaches described thus far. Individual cells and cells within tissues can be detected by use of specific antibodies. Like with FACS, direct immunofluorescence staining takes advantage of direct conjugation of a fluorescent probe to a primary antibody whereas indirect immunofluorescence staining uses a fluorochrome labeled secondary antibody to detect the specific primary antibody. Cell expression of a specific factor can then be studied in detail on a suitable microscope.
Procedure
For frozen tissue sections, remove from −20°C freezer and thaw at room temperature. For adherent cells on cover slips, wash x3 in PBS
Fix the tissue/cells in 4% PFA/PBS for 5–10 minutes and then wash in PBS 3×5 minutes
For tissue sections, clean the slide to remove water around tissue. Use water-resistant pen to circle the tissue sections
Incubate the tissue/cells in 100 μl of blocking solution (PBS + 0.1% Triton X-100 + 2% normal blocking serum) at room temperature for 1 hour
Incubate in 50–100 μl of primary antibody diluted in blocking solution for 1 hour at room temperature or 4°C for overnight. Incubate a negative control in blocking solution without primary antibody
Wash the slide in PBS for 3×10 minutes at room temperature
Incubate the tissue/cells in 50–100 μl of secondary antibody conjugated with fluorophore diluted in blocking solution at room temperature for 45 minutes (protect from light)
Incubate the tissue/cells in 50–100 μl of 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) (5 μM) diluted in PBS for 5 minutes at room temperature to counter stain the nucleus
Wash the slide in PBS for 3×10 minutes at room temperature (protect from light)
Mount the slide/cover slip using with anti-fade mounting medium (e.g., Fluoromount G, Electron Microscopy Sciences), and observe the cells under fluorescence microscope.
Important Note: For double immunofluorescence staining, use two antibodies raised in different species in step 6 and 2 secondary antibodies conjugated with different colors of fluorophores in step 8. The blocking solution should include the sera from both of the animals that secondary antibodies were raised in.
Telomerase activity and telomere length measurement
Several techniques may be used to assess telomere length: Terminal Restriction Fragment (TRF) Southern blot;[82] quantitative-fluorescent in situ hybridization (Q-FISH); RT-PCR; and Flow-FISH. RT- PCR and Flow-FISH overcome the necessity for large amounts of genomic DNA required for Q-FISH. RT-PCR establishes the telomere to single copy gene ration which is proportional to the averaged telomere length within a cell whereas Flow-FISH is adapted from Q-FISH and uses median fluorescence detected by flow cytometry. Gordon et al. describe a TRAP (telomeric repeat amplification protocol) assay to measure telomerase activity in cells, TRF to estimate telomere length, and the anaphase bridge index and the frequency of dicentric chromosomes to detect telomere dysfunction, and readers are encouraged to refer to their paper for these protocols.[82] Protocols to assess telomere length and activity are also described briefly below.
Telomere length
Procedure
Expand the cells to 60–80% confluence
Resuspend and transfer the cells to 1.7 ml microtubes at 1×106 cells/tube
Following the manufacturer's directions, suspend the cells in a hybridization solution containing a FITC-conjugated telomere probe or in a probe-free hybridization solution provided in, for example, a Telomere Assay Kit (Dako Cytomation)
Hybridize each phenotype overnight at room temperature, in duplicate
Wash the cells and incubate the cells with propidium iodide (0.5 μg/ml) to select the cells in G0/G1 of the cell cycle to normalize the data to equivalent genome loads
Analyze the cells in FACS machine to detect the fluorescence intensity of the samples
The fluorescence intensity reflects the relative telomere length (telomere fluorescence per chromosome/genome in the sample with respect to that in control (no fluorescent probe))
Each duplicate forms 1 experiment, an average of 3 experiments is required to determine the significance.
Telomerase activity
Procedure
Lyse 103 cells with lysis buffer provided by the Telomeric Repeat Amplification Protocol (TRAP)-eze telomerase detection kit (Chemicon, CA, USA)
After following the manufacturer's detailed instructions, the PCR products and a 6-base pair incremental ladder are electrophoresed on a 12.5% nondenaturing polyacrylamide gel and visualized by SYBR gold staining (Molecular Probes, Eugene, OR).
Single cell clonogenic assay (adapted from reference 53)
Procedure
Wash 96-well collagen-coated plates with sterile PBS 200 μl per well and replace with 200 μl standard medium (e.g., DMEM + 10% FBS + Pen/Strep)
Resuspend adherent cells using TrypLE (Invitrogen) and strain the cells using a 40 μm filter
Transfer the cells to flow cytometry tubes containing the standard culture medium at 1×106 cells/tube
Sort the cells with a FACS Vantage sorter or equivalent (BD Biosciences) at a rate of 100 events/s; gate the cells based on viability and morphology
Seed each phenotype in triplicate in the 96-well collagen-coated plates with media. Culture the cells at 37°C with 5% CO2-21% O2, and add 100 μl media per well at the top of the old media every 4 days for 12 days
After colonies start to form, remove them using TrypLE (Invitrogen) and expand cells into 6-well plates. Clearly label the individual clones, approximately 20 clones should be generated from each 96-well plate utilized.
SUMMARY
The methodology described in this review should enable a basic identification and characterization of stem and progenitor cells in the pulmonary vasculature. Figure 9 outlines a potential flow of characterization of the putative stem cells that have been discussed in this review. Currently, easy identification is limited due to the lack of exclusively specific identifying markers for different progenitor cells. There is no single marker that can identify a specific stem/progenitor cell; thus investigations still rely upon immunophenotyping of the cell population and sorting of putative stem and progenitor cells prior to confirmation by rigorous functional characterization. Furthermore, the field of stem and progenitor cells in pulmonary vascular disease is continually progressing and becoming more complex. For example, recent progress has been made in defining micro RNAs (miRNAs) capable of modulation vascular cell phenotypes highlighting both a functional and therapeutic significance for small noncoding RNAs in PH.[83] Despite many advancements in the diagnosis and treatment of pulmonary hypertension, it remains a progressive disease with poor prognosis. The role of progenitor cells, be it pathogenic or therapeutic, still remains controversial. All that can be concluded is that preliminary clinical trials utilizing EPC-based therapies in patients with pulmonary hypertension are showing positive effects and indicate that potential therapeutic benefit identified in animal studies may exist.
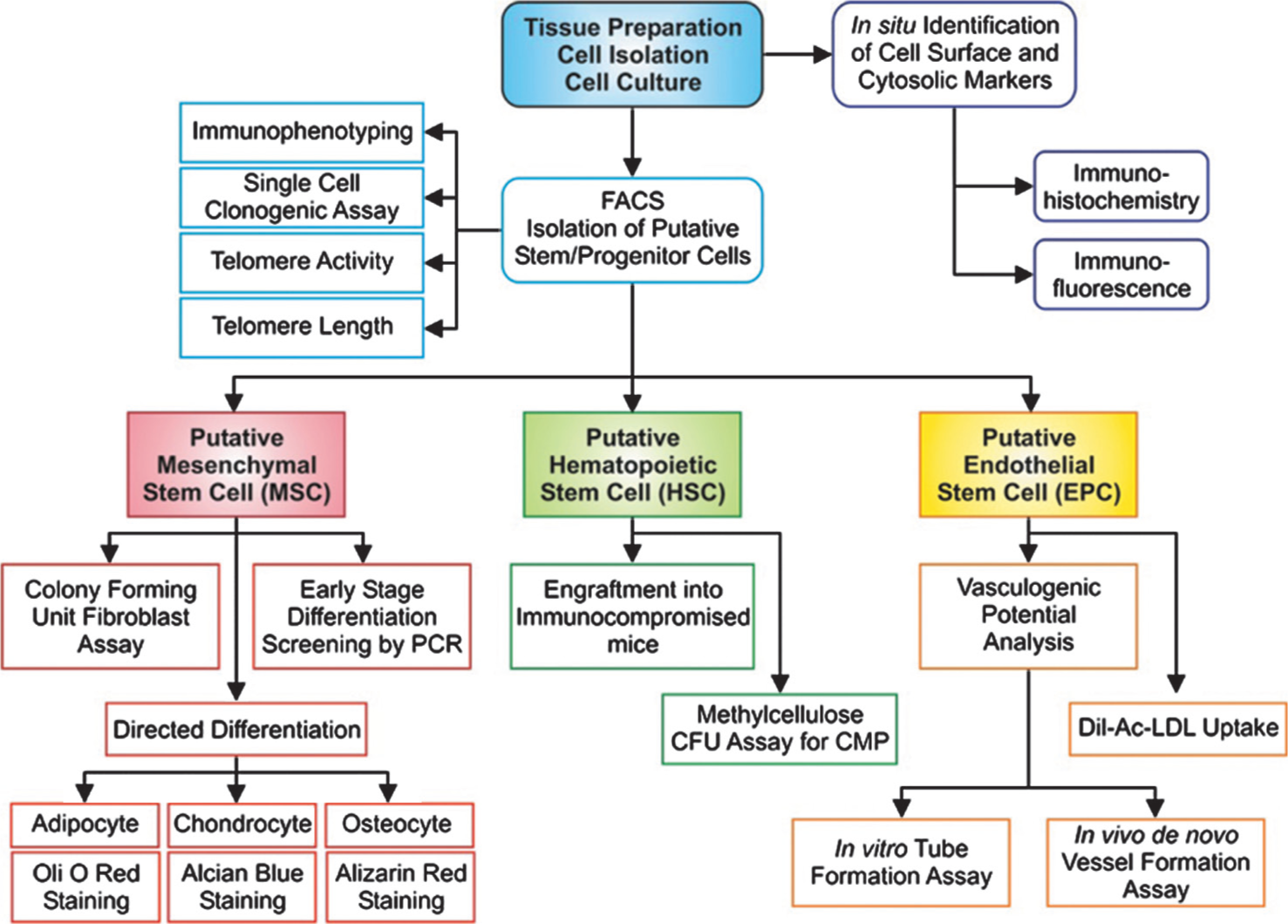
A schematic diagram outlining the key methodology for characterization of putative progenitor cells in the pulmonary vasculature.