
Abstract
Genetically modified mouse models have unparalleled power to determine the mechanisms behind different processes involved in the molecular and physiologic etiology of various classes of human pulmonary hypertension (PH). Processes known to be involved in PH for which there are extensive mouse models available include the following: (1) Regulation of vascular tone through secreted vasoactive factors; (2) regulation of vascular tone through potassium and calcium channels; (3) regulation of vascular remodeling through alteration in metabolic processes, either through alteration in substrate usage or through circulating factors; (4) spontaneous vascular remodeling either before or after development of elevated pulmonary pressures; and (5) models in which changes in tone and remodeling are primarily driven by inflammation. PH development in mice is of necessity faster and with different physiologic ramifications than found in human disease, and so mice make poor models of natural history of PH. However, transgenic mouse models are a perfect tool for studying the processes involved in pulmonary vascular function and disease, and can effectively be used to test interventions designed against particular molecular pathways and processes involved in disease.
Genetically modified mouse models of pulmonary hypertension (PH) have tremendous power in their ability to isolate the function of specific molecular pathways in live animals. However, there are at least two reasons for caution in interpreting the results of PH experiments in mice. First, PH is a disease of diverse etiology, and so no one model can meaningfully capture all variants. Second, even within a subtype of PH, both the timing of the development of disease and physiology in mice is quite distinct from human.
The classification system adopted at the fourth World Symposium on PH held in 2008 in Dana Point, California, split pulmonary hypertension into six broad categories,[1] with each category including several etiologically distinct subcategories. Group 1 includes all causes of pulmonary arterial hypertension (PAH). While much idiopathic PAH (Group 1.1) appears to share molecular etiology with BMPR2-related heritable PAH (Group 1.2.1),[2] these are both clearly distinct in both cause and likely treatments from Schistosomiasis-related PAH (Group 1.4.5) and persistent PH of the newborn (Group 1.5). And all of these are clearly distinct from the most-studied mouse model of PH, chronic exposure to high altitude (Group 3.6).
Moreover, even in focusing on just one of these distinct conditions, there is no mouse model which accurately reproduces the human disease. For instance, while chronic exposure to high altitude produces PH in both mice and humans, mice have a very distinct physiologic response, with far less vascular remodeling than found in larger animals, possibly reflecting the relative lack of adventitia surrounding murine pulmonary vessels.[3] More broadly, human PH develops in free living individuals, likely with multiple genetic contributions, and exposed to numerous environmental stimuli, all of which are missing in mice. Human PH develops over years, rather than the weeks or months used in transgenic mice, and the characteristic pathology is probably the result of years spent with high pulmonary pressures, a feature unachievable in mice.
One thus cannot use a mouse model to replicate the natural history of disease in humans, even if it is possible to start with an identical molecular insult (and the initial insult is still speculative in most forms of PH).
Despite these problems, genetically modified mouse models are uniquely powerful in their ability to study PH-related processes. While the different Dana Point PH groups are in many ways etiologically distinct, they share common processes. Processes in common to almost all PH include altered regulation of tone, remodeling of the vessels through muscularization and intimal lesions, an inflammatory component, and alterations in metabolic state (Fig. 1). In addition, in PH as etiologically distinct as the scleroderma-associated and idiopathic forms, increased estrogenic effect appears to be a risk factor.[4] The relative importance of each of these processes, and the likelihood of each as an initiating event, is distinctive across types of PH, but they are present, and probably at least in part contributory, to all forms. This is the scale of research at which mouse models excel: They enable a reductionist approach to a physiologic process too complicated to study in cell culture, and with invasiveness and control of variables not possible in human patients.
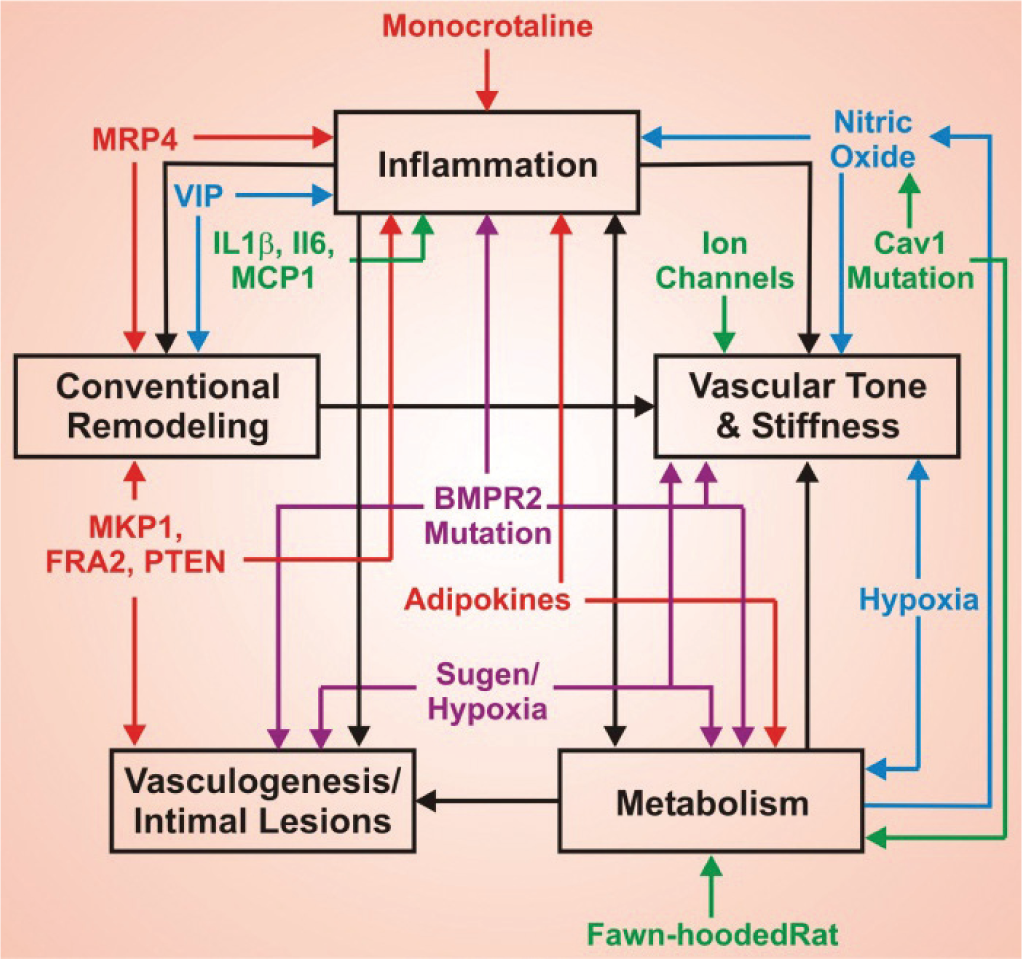
While there are a large number of mouse models of pulmonary arterial hypertension (PAH), different forms of PAH share a number of core processes. These include alterations in metabolism, inflammation, vascular tone and stiffness, vasculogenesis and intimal lesions, and conventional remodeling (muscularization, hypertrophy, adventitial thickening) (bold, black type). There are a tremendous number of PAH models (smaller colored type), which allows detailed examination of these processes, and how they interact to produce disease. There is thus not a “best”’ mouse model of PAH, but rather mouse models specialized in examining different processes of importance to the disease.
MODELS OF REGULATION OF VASCULAR TONE
There are several classes of PH which probably have increased tone as their central causative feature: Vasodilator-responsive idiopathic PAH, persistent pulmonary hypertension of the neonate (PPHN), and PAH related to COPD, for example. However, even for those classes of PH for which increased tone is probably not central, vasodilation will improve quality of life. Every currently approved therapy for idiopathic and heritable PAH uses some class of vasodilator (prostacyclin analogues, endothelin antagonists, and phosphodiesterase-5 inhibitors), despite these drugs clearly not being disease-modifying in most cases.5,6 Study of the regulation of vascular tone is thus relevant to treatment of PH, regardless of whether it is the initiating defect in any particular class of disease.
Studies of vascular tone have focused on secreted factors such as nitric oxide (NO), prostanoids and endothelin (ET), and on ion channels and transporters, and the intracellular components necessary for their proper function.
Hypoxia
Chronic exposure to high altitude (Group 3.6 PH) is nearly perfectly modeled by placing rodents in hypobaric or normobaric hypoxia chambers (there may be differences between the two, but they are subtle). Typically, 10% or 12% effective O2 is used, usually for three to five weeks. This results in a reliable increase in RVSP of roughly 10 mmHg in mice (and more in rats), with attendant right ventricular hypertrophy and remodeling of the pulmonary vasculature. There is a vast body of literature on hypoxic PH in rodents and cattle, recently reviewed,[3] which suggest that while acute hypoxia results in acute vasoconstriction, the long term effect is substantially reliant on and can be blocked by interventions against inflammatory, metabolic, and proliferative processes as well.[3]
The main advantage to hypoxia is its simplicity and reliability. This is probably why it is by far the most common model of PH found in the literature. It is also a perfect model of WHO Group 3.6 PH. Unfortunately, it is often used as a stand-in for all forms of PH, for many of which it is probably molecularly irrelevant. Further, because it is dependent on not just vasoreactivity, but also inflammation, metabolism, and proliferation, it is far less amenable to a reductionist approach than are genetic models.
Nitric oxide-associated genes
Nitric oxide is generated from L-arginine by three nitric oxide synthase (NOS) enzymes, neuronal NOS (nNOS, NOS1), endothelial NOS (eNOS, NOS3), and inducible NOS (iNOS, NOS2). All three isoforms are present in the lung, although nNOS does not appear to have a role in modulating pulmonary vascular tone. eNOS is most responsible for endothelium-dependent vasodilation, and both iNOS and eNOS set basal tone.[7]
Both knockout and transgenic overexpression mice exist for all three isoforms; the primary phenotype in each was described more than 10 years ago. eNOS knockout mice have systemic hypertension, 8 while transgenic mice overexpressing eNOS in the vascular wall have systemic hypotension. 9 Inducible NOS (iNOS) knockout mice had enhanced inflammatory responses to LPS[10] and in allergen models; its role in the allergen models but not in LPS models requires intact nNOS.[11]
Dysregulation of nitric oxide metabolism is probably particularly relevant to research in the field of persistent pulmonary hypertension of the newborn (PPHN). Active areas of research include increasing NO downstream effect using phosphodiesterase inhibitors,[12] use of tetrahydrobiopterin (BH4) as a catalyst for production of NO,[13] and improving transport of NO precursor, citrulline.[14] Hph-1 mice, which have a 90% loss of activity of GTP-cyclohydrolase 1, a protein required for enzymatic synthesis of BH4, have constitutive pulmonary-specific hypertension (RVSP increased by ~10 mmHg), with decreased BH4 and NO[15] and higher ROS.[16] The increase in normoxic RVSP in BH4 deficient mice is comparable to that of eNOS knockout mice.[7] In combination, these data imply that pulmonary specificity can be produced by precursor metabolism and transport rather than through the nitric oxide synthases directly. To date, there are no transgenic or knockout mice built to examine the neutral amino acid transporters involved in citrulline transport.
In addition to regulating endothelium-mediated vasodilation, nitric oxide is involved in a variety of processes, including bronchodilation, anti-adherence, antimitogenic, and anti-inflammatory properties,[17] as well as playing an important part in angiogenesis.[18] eNOS is essential for neovascularization[19] and for fetal lung development.[20]
Finally, nitric oxide uncoupling leads to increased oxidative stress, and to nitrosylation defects which can deregulate other signaling pathways, discussed in combination with Cav1 defects under metabolic stresses, below.
Prostanoids
Prostanoids are produced by the cyclooxygenase pathway from arachidonic acid via a prostaglandin H2 intermediate. There are multiple prostanoids: In addition to prostacyclin, there is prostaglandin D2, E2, and F2α, and thromboxane A2, each produced by a different enzyme acting on PGH2 and each with a different receptor (reviewed in Helliwell 2004[21].
Prostacyclin, produced by prostacyclin synthase and acting through the prostaglandin I receptor (Ptgir), is a vasodilator, whereas prostaglandin E2 and thromboxane are constrictors. However, prostacyclin does not seem to normally regulate pulmonary vascular tone in any animal.[22,23] Ptgir deficient mice are normotensive, but with increased susceptibility to thrombosis and reduced inflammatory responses.[24]
The combination of these data suggests that the primary mechanism of action of prostacyclin is quite different than regulation of tone, as the PH community usually assumes; the topic will be discussed again in the section on inflammatory processes. Because prostacyclin was developed as a therapy for PH before the era in which transgenic mice were common, careful studies of gene modified mice were never performed to determine its mechanism. However, because of the specificity of both receptor and synthesizing enzymes, it is an area extremely amenable to genetic investigation.
Endothelin
Endothelin-1 (ET1, EDN1) is a 21 amino acid peptide, expressed primarily in endothelial cells, made from a 213 amino acid propeptide by endothelin converting enzyme (ECE). ET1 is principally known as an extremely potent vasoconstrictor, acting through two g-protein coupled receptors, endothelin receptor A (ET-A, EDNRA) and endothelin receptor B (ET-B, EDNRB). Bosentan, a current treatment for PAH, is an endothelin receptor blocker. Knockout mice for ET1, ECE, and both receptors have been made.
ET1−/− mice die of respiratory failure at birth, with additional craniofacial developmental abnormalities.[25] ET1+/− mice have slightly elevated systemic pressures[25] and are hyper-responsive to methacholine in asthma models.[26] Conversely, overexpression of ET1 in transgenic mice does not cause elevated pressure, but does lead to hypertrophy, increased oxidant stress,[27] and vascular inflammation.[28] It appears, then, that with either constitutive overexpression or deletion of ET1, a new setpoint is reached which prevents a role in vascular tone. However, both overexpression and deletion still have important roles in regulating development, angiogenesis, and inflammation.
Endothelin receptor A knockouts have defects in craniofacial development that mimics ET1 knockouts, in addition to defects in the cardiovascular outflow tract.[29] By contrast, endothelin receptor B knockouts have a phenotype dominated by colon and coat developmental abnormalities.[30] Knockout of ECE-1 produces a phenotype indistinguishable from either ET1 or EDNRA knockout.[31]
While both endothelin receptors apparently have important roles in regulating different populations of neural crest cells, the phenotype in knockouts is not particularly relevant to PAH. It seems likely that conditional and tissue-specific abrogation would be needed to produce a mouse with a phenotype more relevant to PAH, by avoiding developmental defects. ET1 has important roles in regulating many PAH-related activities, including inflammation and angiogenesis, and so conditional expression or knockout of ET1 and its receptors would likely add significantly to our understanding of details of its function.
GENETIC MODELS INVESTIGATINGION CHANNELS
The operation and interaction between various potassium and calcium channels, their internal stores, and regulation of sensitivity, is bewilderingly complex, and an active area of research (covered recently in a comprehensive fashion by the proceedings of the 2008 Grover Conference[32].
Genetically altered mouse models are particularly useful for studies of ion channels in vascular smooth muscle cells (VSMCs) and pulmonary arterial hypertension (PAH) because of lack of a specific blocker that entirely blocks its targeted channel without direct or secondary effects on any other channels. This review focuses on transgenic and knockout mice that have been used to identify the function of potassium and calcium channels in VSMCs and in the development of pulmonary hypertension.
Potassium channels
K+ channels are the key determinant of the resting membrane potential in SMCs and regulate [Ca2+]i in pulmonary arterial myocytes that in turn regulate pulmonary vascular tone and remodeling.[33] In mammals, K+ channels are divided into five major classes according to their transmembrane segments (TMSs): (1) Calcium activated K+ channels (BKCa); (2) voltage gated K+ channels (KV); (3) ATP sensitive K+ (KATP); (4) inwardly rectifying K+ channels (Kir); and (5) tandem pore domain K+ channels (K2P). Kir and KATP channels consist of two TMSs domains and one P domain. KV and BKCa channels are characterized by six or seven TMSs and both are sensitive to voltage changes. Four TMSs and two P domains make up K2P channels that are open at rest and are qualified as leak or background K+ channels. All of these five major classes of potassium channels have been identified in VSMC, and PAH is associated with a loss of K+ channel expression and activity, both in animal models and human patients. Thus, K+ channels appear to be preferentially expressed in pulmonary arteries and constitute potential therapeutic targets for the treatment of tone-related PH.
In the SWAP mouse model, which is functionally a Kv1.5 knockout mouse, hypoxic pulmonary vasoconstriction was reduced significantly compared to wild type mice.[34] Although, pulmonary hypertension did not occur as expected in Kv1.5 knockout mice, the PASMCs from SWAP mice were slightly more depolarized and had less hypoxia-sensitive K+ current (IK) than did wild type cells. 34 Both mice overexpressing Kv1.5 and mice adenovirally overexpressing a phosphorylation-deficient survivin mutant have been reported to reduce PAH resulting from restoring of Kv1.5 expression and increased KV channel current.[35,36] These studies raised the possibility of gene therapy targeting KV channels for the treatment of PAH.
In VSMCs, BK channels are comprised of an alpha pore (BKα) and one of the four beta subunits (BKβ1) that modulate Ca2+ sensitivity.[37] Iberiotoxin, BK channels specific blocker, can induce marked membrane depolarization and vasoconstriction,[38] and inhibit the actions of a variety of smooth muscle relaxants.[39] These studies implicated BK as a negative feedback control element to regulate membrane depolarization and vasoconstriction in smooth muscle. Brenner et al. first reported that BKβ1 gene deletion leads to a decrease in the calcium sensitivity of BK channels, a reduction in functional coupling of calcium sparks to BK channel activation, and increases in arterial tone and blood pressure.[40] Subsequently, some studies have also found hypertension in BKβ1 KO mice.[41–44] In concert with BKβ1 KO studies, Sausbier et al. showed that deletion of the BK channel α subunit also leads to a small but significant elevation of blood pressure resulting from decreased serum K+ levels and increased vascular tone.[45] However, Xu et al. reported that BKβ1 knockout mice are not hypertensive and the higher pressure observed in the previous studies may be due to shortly and indirectly pressure measurements, different age and genetic background, or restraint-induced stress and sympathetically mediated vasoconstriction.[46] Although, BK channel deletion lead to no or mild elevation of blood pressure in normal conditions, it is very possible that BK channels play a role in abnormal situations, such as PAH.[46] Therefore, the contribution of BK channels function to regulation of vascular tone and hypertension still requires further research.
K2p channels have been shown to conduct leak K+ and have emerged as prime candidates for controlling the resting membrane potential in PASMCs.[47] Knockdown of the acid sensitive, tandem-pore domain K+ channel type-1 (TASK-1) was shown to mediate the depolarizing effects of hypoxia,[48] and Endothelin-1 also mediates depolarization in human PASMCs via inhibition of TASK-1.[49] However, Manoury et al. found that the pulmonary arteries from TASK-1 knockout mice do not display any different properties compared to wild type mice.[50] Their results imply that TASK-1 channels do not play role in the regulation of either the resting or the agonist induced contractile of mouse PA and raise the question of whether mice can be used to appropriately model this aspect of human pulmonary vascular physiology and pathology.[50]
Calcium channels
In PASMCs, elevation of Ca2+ concentration has been suggested as a major trigger for pulmonary vasoconstriction, hypertension, and VSMC cell growth, proliferation, and migration.[51,52] Increase of [Ca2+]i results from Ca2+ release from intracellular organelles or Ca2+ entry from the extracellular space that are mediated by voltage gated Ca2+ channels, receptor operated Ca2+ entry channels (ROCE), and store operated Ca2+ channels (SOCE).[53] Several Ca2+ channels knockout mice have been generated which could tell us more about the physiological role of these Ca2+ channels in VSMCs and in the PAH.
By crossing floxed Cav1.2 mice with SM-Cre ERT2 mice to overcome embryonic lethality, Moosmang et al. generated tamoxifen-induce SMC conditional Cav 1.2 knockout mice. The spatially and temporally controllable knockout mice have normal body weight, breed normally, and are indistinguishable from control littermates before tamoxifen injection.[54] With inactivation of Cav1.2 by tamoxifen injection, mice show reduced mean arterial blood pressure, and development of myogenic tone in response to intravascular pressure was absent. These mouse studies support the theory that Cav1.2 Ca2+ channels are essential to the regulation of blood pressure, vascular contractility, and myogenic tone,[54] and also suggested that L type ca2+ channels could be the sensors coupling membrane depolarization to SR Ca2+ release.[55]
Transient receptor potential (TRP) proteins, including TRPC, TRPM, and TRPV, are widely expressed in VSMCs and are implicated in SMCs contraction and proliferation and pulmonary hypertension. TRPC1 knockout mice showed no significant differences in cation currents and no change in store operated cation influx induced by thapsigargin, inositol 1, 4, 5 trisphosphate, and cyclopiazonic acid.[56] TRPC4 knockout resulted in impairment of store operated Ca2+ entry, disturbed endothelium dependent vasorelaxation, increase of microvascular permeability and impaired contraction in SMCs.[57–59] TRPC6 knockout mice showed an elevation of blood pressure and enhanced agonist-induced contractility of isolated aortic rings and cerebral arteries.[60] SMCs from TRPC6 knockout mice have increased contractility and higher basal cation entry resulting from upregulated TRPC3 type channels expression and activity. Further studies on the knockout mice found that TRPC6 activity is essential for acute hypoxic pulmonary vasoconstriction but is not involved in chronic hypoxia induced pulmonary hypertension.[61] Interestingly, G Welsh et al. reported that TRPC6 knockdown by antisense oligodeoxynucleotides decreased VSMCs depolarization and constriction induced by elevated pressure.[62] The differences in the results from knockdown and knockout might be due to the efficiency of gene suppression in the knockdown model.[63] The results from TRPV4 knockout mice supported that TRPV4 channels are critically involved in the vasodilatation of mesenteric arteries in response to endothelial derived factors.[64] Some other TRP channels knockout mice also have been generated, such as TPRV1,[65] TRPM2,[66] and their expressions have been characterized in PASMC.[67] However, very little is known about how these channels contribute to PAH in vivo. Knockdown of members of SOCE channels, STIM1, and Orai 1, inhibited synthetic VSMC proliferation and migration, whereas STEM2, Orai2, and Orai3 knockdown had no effect.[68] Their precise function in PAH is not known because of no knockout mouse has been made.
Based on data derived from studies of pharmacological blockers or inhibitors, some ion channels have been proposed to play critical roles in the development of PAH. However, knockout mice did not show the phenotypic changes in PAH thathad been predicted. It is possible that deletion in one of these ion channels may be compensated by the expression of other ion channels. Therefore, it is important to make certain that any phenotypic changes or lack thereof result from the target gene knockout, not from disruption or compensation of or by other genes. Despite these caveats, with appropriate controls, ion channel transgenic and knockout mice allow us to identify the function of ion channels of interest in a complex situation at different levels from animal physiology and pathophysiology, to cell biology and molecular biochemistry.
METABOLIC AND OXIDATIVE STRESSES IN RODENT MODELS OF PULMONARY ARTERIAL HYPERTENSION
Altered energy metabolism and metabolic reprogramming are increasingly recognized as highly correlated with and potentially causal in the pathogenesis of pulmonary arterial hypertension (Fig. 2).[69] Multiple facets of energy metabolism have been found to be dysregulated in several different animal models of PAH, and these metabolic abnormalities have been found to be reflected in humans with PAH. Modulation of energy metabolism is emerging as a promising new therapeutic strategy for PAH. The utility of modulation of individual metabolic pathways for PAH in humans relies upon the validity of the corresponding findings in animals. The various facets of metabolic dysregulation in rodent models of PAH are discussed below.
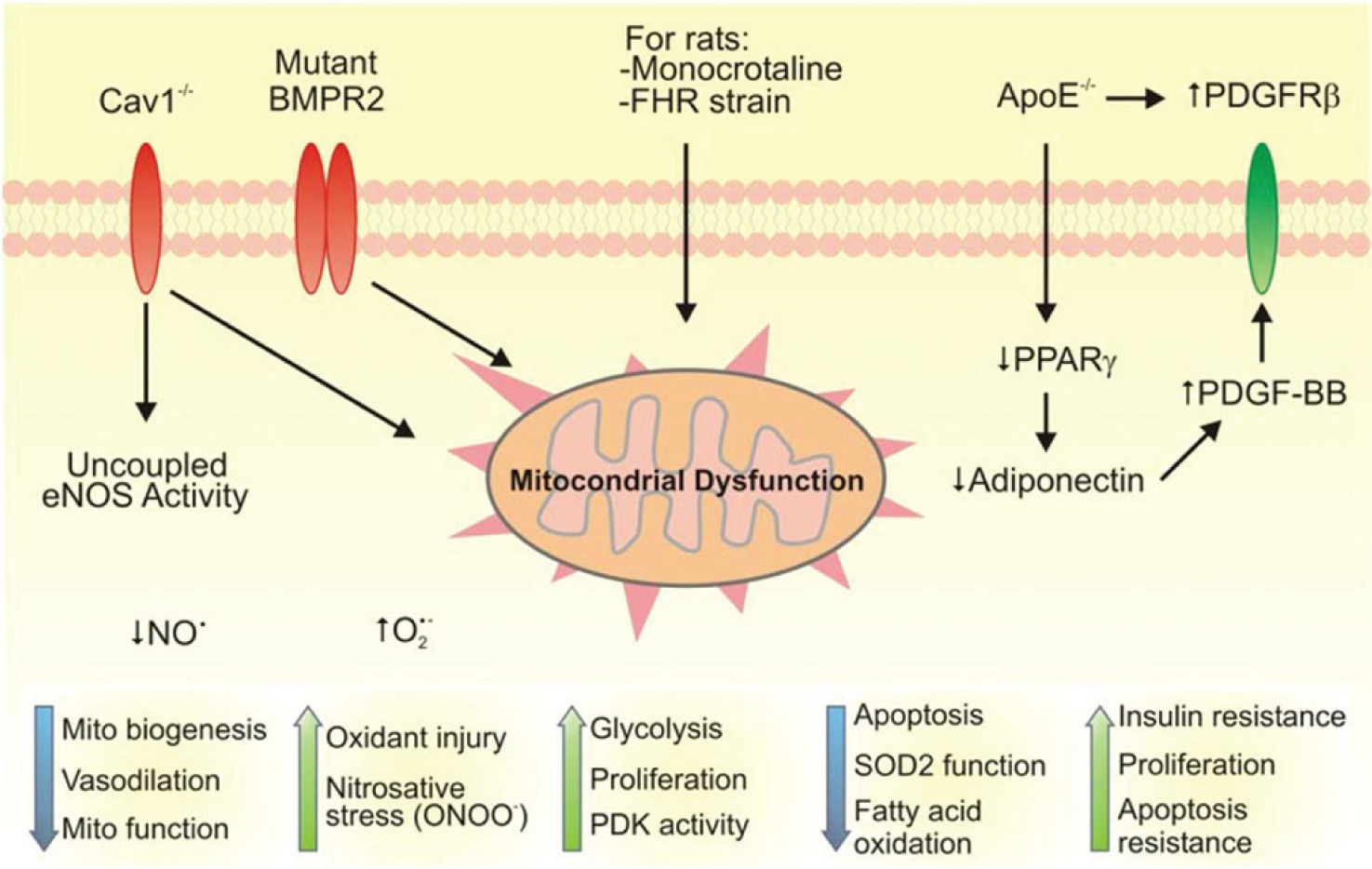
Graphical summary of the dysfunctional oxidative and metabolic pathways thought to be pathogenic in various models of PAH. Across the top are the various perturbations/treatments that characterize the models (e.g., Cav1 −/−, mutant BMPR2, etc.). Beneath each of these are the pathways thought to be central to the molecular pathogenesis of each model (e.g., mitochondrial dysfunction for mutant BMPR2, monocrotaline rats, and Fawn hooded rats). At the bottom are the final common pathways that are thought to lead to the PAH phenotype. Cav1 = caveolin 1; eNOS = endothelial nitric oxide synthase; NO = nitric oxide; O2- = superoxide anion; ONOO- = peroxynitrite; BMPR2 = bone morphogenetic protein receptor type 2; FHR = Fawn hooded rat, PDK = pyruvate dehydrogenase kinase, ApoE = apolipoprotein E, PPARgamma = peroxisome proliferator activated receptor gamma, PDGF = platelet-derived growth factor, PDGFR = PDGF receptor.
The fawn-hooded rat
One of the first rodent models in which metabolic dysfunction was recognized as a key contributor to PAH pathogenesis is the Fawn-hooded rat (FHR). The earliest published report of any sort of metabolic abnormality in the FHR strain was in 1975, when Raymond and Dodds noted that the FHR strain had a significantly lower serum glucose than Wistar counterparts.[70] At that time, the FHR strain was used for studies of coagulation dysfunction, as the strain has intrinsic prolongation of the partial thromboplastin time, as well as reductions in platelet serotonin content and release. In the mid-1980s the strain was found to develop age-related systemic hypertension and proteinuria and thus became a model for the study of systemic hypertension and its complications.[71–73] At this same time, Kuijpers and de Jong noted that the FHR developed pulmonary congestion, evidence of vasculopathic changes in multiple vascular beds, and marked myocardial fibrosis and hypertrophy.[72] This was the earliest recognition that the FHR could be a useful model for studying pulmonary vascular disease.
Abnormalities in serotonin storage in both platelets and the central nervous system in FHR were recognized before any specific defects in energy metabolism, though hyperfunction of the hypothalamic-pituitary-adrenal axis with resultant hypercortisolemia had been described/[74–79]
The earliest description of specific energy metabolism abnormalities in the FHR was published by Bonnet and colleagues in 2006.[80] This study was based upon previous work showing that the metabolic modulator dichloroacetate reversed the resistance to apoptosis seen in pulmonary artery smooth muscle cells (PASMCs) in rodent models of hypoxic pulmonary hypertension and monocrotaline-induced PAH, and that it did so through modulation of hypoxia-inducible factor signaling and activity of the redox-sensitive potassium channel Kv1.5.[81,82] In PASMCs from the FHR, marked mitochondrial abnormalities were noted, including decreased expression of complex I of the electron transport chain, hyperpolarization of the mitochondrial membrane, decreased expression of SOD2, and disruption of the normal mitochondrial reticular network. Rehman and Archer then went on to show that PASMCs from the FHR exhibited a shift toward glycolytic metabolism even in the presence of adequate oxygen concentrations to permit efficient oxidative phosphorylation.[83] Metabolic reprogramming toward aerobic glycolysis has long been recognized as a feature of cancer cells, and part of the beneficial effect of dichloroacetate has been shown to be related to its ability to inhibit pyruvate dehydrogenase kinase and to shift cellular energy metabolism away from glycolysis and toward glucose oxidation.[84–86]
The metabolic abnormalities in the FHR model overlap not only with many of those described for hypoxic pulmonary hypertension, but emerging data have shown overlap with other animal models of PAH (see below) and with human PAH.[87,88] However, it should be noted that the FHR is most accurately described as an animal strain that develops a variety of organ system dysfunctions, of which PAH is only one facet. As mentioned above, the FHR model was first utilized as a model of systemic hypertension, hypertensive renal disease, and platelet and coagulation dysfunction. These are not features that are commonly associated with PAH. Indeed, it has been suggested that the FHR may in fact be best described as an animal model for Hermansky-Pudlak syndrome, which can involve pulmonary hypertension as part of the disease complex but which is usually secondary pulmonary hypertension related to pulmonary fibrosis.[80,89] Additionally, few if any studies of spontaneous PAH in the FHR have looked closely at the left ventricle to confirm that what is being studied is not in fact WHO Group II pulmonary hypertension. A recent investigation of left ventricular dysfunction in type 2 diabetes mellitus has shown that the presence of mitochondrial DNA from a Fawn-hooded rat causes left ventricular remodeling, mitochondrial and myofibril disorganization, and dysfunction of complexes I and IV in the electron transport chain. These changes are not present in a rat with an identical nuclear genome but with a mitochondrial genome from a Wistar rat background.[90]
Knockout of murine apolipoprotein E, PPAR-gamma, or adiponectin
Early studies examining pulmonary vascular remodeling in both chronic hypoxic pulmonary hypertension, as well as monocrotaline-induced pulmonary hypertension pointed to dysregulation of the insulin signaling machinery (e.g., decreased peroxisome proliferation activating receptor gamma [PPAR-γ] signaling, increased insulin-like growth factor signaling), as well as increases in platelet-derived growth factor (PDGF) signaling.[91–95]Additionally, lungs from patients with idiopathic PAH were found to have decreased levels of transcripts for PPAR-γ, as well as for apolipoprotein E (apo E).[96,97] These findings were brought together in 2007 by Hansmann and colleagues, who showed that apo E −/− mice fed a high-fat diet to further drive insulin resistance developed PAH as measured by elevated right ventricular systolic pressure (RVSP).[98] However, only male knockout mice developed RV hypertrophy, muscularization of the pulmonary arteries, insulin resistance that correlated with severity of RVSP elevation, and decreased plasma levels of adiponectin. In male knockout mice treated with the PPAR-γ agonist rosiglitazone, all of these findings substantially normalized.
Hansmann et al. proposed a pathogenic mechanism for PAH in which loss of apo E decreases internalization of the PDGF rececptor β, and decreased PPAR-γ signaling leads to insulin resistance and to decreased sequestration of PDGF-BB by adiponectin. This group has gone on to show that targeted deletion of PPAR-γ in smooth muscle cells or in endothelial cells results in spontaneous PAH.[99,100] and Summer and colleagues have shown that knockout of adiponectin also results in the development of PAH.[101] Emerging data have also shown that insulin resistance and glucose intolerance out of proportion to body mass index (BMI) are more prominent in PAH patients compared to age- and BMI-matched controls.[102,103] Based upon these studies, therapies targeted at insulin sensitization and/or PPAR-γ activation seem promising,[104] although there are currently no active trials of thiazolidinediones in PAH, the most recent having been terminated in July 2011 due to difficulty in recruiting eligible subjects.
The models described above have some significant concordances with human PAH, though there are notable deviations worthy of discussion. Most notably, in the apo E −/− mice, males showed the most pronounced PAH phenotype, whereas in human PAH females are much more significantly affected than males. The models that have so elegantly probed this pathway also all seem to converge on PDGF as a key growth factor exhibiting increased signaling and functioning as a pathologic mitogen. Indeed, Hansmann and colleagues have suggested that the pathogenic effects of loss of growth inhibitory signaling from bone morphogenetic protein receptor type 2 (BMPR2) in PAH are mediated through PPAR-γ and apo E, thus indirectly implicating PDGF.[99] However, despite implication of pathological PDGF signaling, and despite evidence that inhibition of PDGF may be beneficial in PAH,[105–108] no group has shown that deletion of PDGF or its receptor in an animal model prevents or reverses PAH. This may be relevant, given that the first completed trial of imatinib for PAH (Imatinib in Pulmonary Arterial Hypertension, a Randomized Efficacy Study— IMPRES) found an improvement in the imatinib-treated group in measured pulmonary hemodynamics and in exercise tolerance measured by 6-Minute Walk Distance, but no difference from placebo in time to clinical worsening (defined as death, hospitalization for PAH, a worsening of WHO functional class, or a decrease of 15% or more in 6MWD).[109] In addition, in both the initial IMPRES study and in interim results reported from a planned three-year extension study, serious adverse events have been more common in the imatinib treated group, including subdural bleeding.[110]
Monocrotaline-treated rat
Treatment of rats with the toxic alkaloid monocrotaline is perhaps the most widely used animal model of what is considered to be WHO Group I pulmonary hypertension. Monocrotaline was first recognized as a hepatotoxin as early as the 1940s,[111] with reports of its effects on the pulmonary vasculature and myocardium published in the 1960s.[112–114] Early evidence suggested that part of the systemic toxicity of monocrotaline involved alterations of cellular metabolism and oxygen consumption.[115] Monocrotaline's toxicity toward cardiac mitochondria was recognized by Guarnieri and Muscari in 1988, who found reduction in complex I, II, and IV activities as well as increased superoxide production in cardiac mitochondria following high-dose (105 mg/kg) monocrotaline treatment.[116] Monocrotaline has been shown to upregulate aerobic glycolysis in cardiomyocytes and in hepatocytes.[117,119] Compounds thought to inhibit fatty acid oxidation and restore more normal glucose oxidation (e.g., trimetazidine) have been shown to be beneficial in monocrotaline-induced PAH,[116,120] as have pyruvate dehydrogenase kinase inhibitors such as dichloroacetate.[82,117]
Despite important correspondences between the metabolic defects in human PAH and in the monocrotaline-treated rat, there are important differences that bear consideration, particularly since this is the most commonly used model of PAH that does not involve hypoxia (hypoxia being a model of WHO Group III pulmonary hypertension).[121] Monocrotaline is most accurately described as a model of multiorgan system toxicity, of which the pulmonary vascular and cardiac toxicities are only a part. Monocrotaline has also been shown to cause pulmonary fibrosis, hepatic injury, immunotoxicity, and DNA crosslinking.[122–131] More importantly, though several different metabolic modulators have been shown to be beneficial in monocrotaline-induced PAH, and though these may indeed be beneficial in human PAH, the pathologic changes induced by monocrotaline have been shown to be reversed by a large number of agents that have not been beneficial in human disease.[132–136]
Endothelial nitric oxide synthase and caveolin-1
A feature of the pathobiology of PAH is decreased bioavailable nitric oxide.[137] Loss of a key mediator of vasodilatory tone is a mechanistically pleasing argument for the development of PAH. However, early data from murine knockout models of endothelial nitric oxide synthase (eNOS) presented somewhat conflicting data, with exaggeration of stimulated vasoconstriction observed in the pulmonary circulation but in the absence of spontaneous development of PAH.[138,139] This apparent conundrum was at least partially resolved by the finding that eNOS uncoupling contributes to PAH; that this uncoupling leads to increased generation of superoxide, peroxynitrite, and nitration of protein kinase G; and that supplementation with tetrahydrobiopterin (a required cofactor for eNOS function) partially ameliorates disease.[16,140–145] Further support for eNOS uncoupling in PAH comes from the finding that caveolin-1 knockout (Cav1 −/−) mice exhibit increased eNOS activity but develop PAH that is dependent upon eNOS, and that loss of eNOS function through pharmacologic inhibition or genetic deletion prevents development of disease in the Cav1 −/− mouse.[142,146–150]
Though there is good evidence to suggest that oxidative/nitrosative stress is an important pathogenic mechanism for PAH in the Cav1 −/− mouse (see below), metabolic stress may also be an important and as yet underappreciated feature of this model. Decreased nitric oxide bioavailability has been directly linked to decreased mitochondrial biogenesis and mitochondrial dysfunction in PAH.[88] In hepatocytes and mesenchymal stromal cells, loss of Cav1 promotes a shift to a glycolytic phenotype, much like that which has been described in PAH.[151,152] Caveolin-1 may also play an important regulatory role in fatty acid metabolism and the efficiency of mitochondrial coupling, as the Cav1 −/− mouse exhibits impaired nonshivering thermogenesis.[153] Loss of Cav1 results in mitochondrial dysfunction related to accumulation of cholesterol in mitochondrial membranes.[154,155] Finally, there is evidence for an association between loss of Cav1 and a hyperproliferative phenotype that is dependent upon loss of function of SOD2, similar to the hyperproliferative phenotype observed in other models of PAH.[156,157]
BMPR2 mutations
Since the identification of mutations in BMPR2 as the causative genetic lesion in approximately 80% of cases of heritable PAH and approximately 20% of cases of IPAH, investigators have been searching for the pathogenic molecular pathways that lead from the gene to the disease.[158] In endothelial cells, BMPR2 signaling has been implicated upstream of PPAR-γ/β-catenin-mediated transcriptional activation of apelin, the loss of which is proposed to drive the development of PAH through the mechanisms discussed above.[159,160] As mentioned above, BMPR2 signaling has also been shown to interact with PPAR-γ signaling in PASMCs to exert an anti-proliferative effect.[99] BMPR2 has also been proposed as an activator for eNOS, with mutant receptor isoforms failing to activate eNOS and perhaps contributing to vasoconstriction as well as impaired mitochondrial biogenesis.[161] Dysruption of normal cytoskeletal regulation and oxidative stress have both been implicated as important pathogenic mechanisms downstream from mutant BMPR2.[2,162,163]
Mounting evidence suggests that metabolic reprogramming is also a key pathogenic mechanism downstream from BMPR2 mutations. Analysis of multiple gene expression arrays from animals or humans harboring BMPR2 mutations have found metabolic genes to be one of the largest gene ontology groups that is differentially regulated in the mutant condition.[163–165] Very recently, mice universally expressing disease-causing BMPR2 mutations have been shown to develop cardiac steatosis and right ventricular failure,[166] and endothelial cells expressing mutant BMPR2 and subjected to a combined transcriptomic/metabolomic analysis have been shown to exhibit widespread metabolic abnormalities (e.g., impaired Krebs cycle function, impaired glutamine, and branched chain amino acid metabolism, decreased fatty acid oxidation, increased glycolysis) that were reflected in patients with heritable or idiopathic PAH.[167]
Oxidative stress/signaling in PAH
Oxidative stress has been implicated as a pathogenic process in virtually every cellular, animal, and human model of pulmonary hypertension. This topic has been relatively recently reviewed in the particular setting of PAH.[168] Sources of reactive oxygen species in PAH include mitochondria, NADPH oxidase, xanthine oxidase, uncoupled eNOS, and activated inflammatory cells.[157,169–172] The underlying assumption in many studies is that overproduction of reactive oxygen species results in deleterious covalent modification of proteins, lipids, and nucleic acids.[162,173,174] The logical hypotheses to follow from this line of reasoning is that enhancement of oxidative stress ought to exacerbate the PAH phenotype, and reduction of free radical production with antioxidant treatment (supplementation of small molecule or enzymatic antioxidants) should ameliorate disease. Enhancement of oxidative stress by knocking out antioxidant enzymes such as extracellular superoxide dismutase (EC-SOD) or application of a pro-oxidant stimulus such as monocrotaline, have been shown to contribute to the PAH phenotype.[175–177] Conversely, there are at least some data that point to a beneficial effect of antioxidants in ameliorating experimental PAH,[178–181] though antioxidant trials in humans for a variety of diseases have thus far been fairly disappointing.
Particular attention must be paid to precision of terminology when planning and reporting investigations examining oxygen radical flux and “reactive oxygen species,” particularly where superoxide anion and hydrogen peroxide are concerned. Often, the implicit assumption is that increased production of ROS is deleterious and is thus synonymous with “oxidative stress.” Terminology that distinguishes between the unregulated and deleterious covalent modification of macromolecules and the generation of regulated signaling species/second messengers has implications for investigation of underlying pathogenesis and subsequent therapeutic design. We propose “oxidant injury” and “oxidant signaling” to distinguish between the two situations. For example, hydrogen peroxide production in PAH has been shown to be a stimulus for both oxidant injury with resultant damage to macromolecules, and for oxidant signaling with restoration of normal apoptosis.[157,182,183] Studies that quantify ROS that can function both as injurious species and as second messengers (e.g., superoxide anion, hydrogen peroxide) should undertake additional experiments to assess oxidant injury versus oxidant signaling. In settings where oxidant injury is the predominant pathogenic mechanism, antioxidant therapy may be of greater benefit. If disrupted oxidant signaling is the predominant pathogenic mechanism, efforts to modulate the production and the targets of the oxidant second messenger (such as has been done with nitric oxide, a potent free radical second messenger) may be more fruitful.
Conclusions as to significance of metabolic factors in pulmonary hypertension
Metabolic and oxidative stresses have been implicated in many different models of PH as well as in humans with disease. Some of the metabolic abnormalities appear to be common to a number of models, though these investigations are far from complete. It remains to be seen whether metabolic modulation in humans will be as effective as it has been in animal models. Whether metabolic reprogramming is a primary pathogenic mechanism or is secondary to another more fundamental molecular lesion is as yet unclear. Moreover, the key regulators that serve as the conductors for the symphony of metabolic derangements in PAH are almost entirely unknown, and it seems unlikely that HIF is acting alone in this regard. Finally, opportunities exist for the creation and investigation of animal models of PAH that stem from and focus on metabolic abnormalities. For example, a murine model of mutation of the NFU1 gene, which in humans causes pulmonary hypertension and profound disruption of mitochondrial function by way of abnormal maturation of iron-sulfur cluster proteins, would help to solidify the causal connection between metabolism and PAH and would allow for more in-depth molecular analyses.[184] The importance of redox biology in PAH is well established in multiple animal models and in humans with disease. Going forward, a clearer distinction between oxidant injury and oxidant signaling (both as distinct phenomena from “oxidative stress”) may help in the translation of findings in animal models to novel therapeutic strategies in patients.
SPONTANEOUS REMODELING OF PULMONARY VASCULATURE IN TRANSGENIC MICE
Several experimental mouse models of PH have been developed by manipulating the dose of candidate genes shown to be involved in the experimental or clinical PH. These genetic approaches used either gene overexpression or knockout strategies. Although, mice develop chronic hypoxia-induced PH, the pathophysiology resulting from such exposure to hypoxia is substantially different from that seen in the human disease. An additional important point to consider regarding generation of mouse models of PH is that the responses of mice to hypoxia are strain-specific and the interpretation of the pathobiological data from such mouse models must include careful consideration of the genetic background of the animals studied.[185] Response to chronic hypoxia in mice is unlike that of humans, because while mice share with humans the symptomatic elevation of pulmonary artery blood pressure, they display only a minimal degree of vascular remodeling.[186–188] The most common findings in mice in response to hypoxia exposure are muscularization of previously nonmuscularized vessels and a minimal medial thickening of muscular resistance vessels. Only brief increases in SMC proliferation are observed and some reports demonstrate an overall decrease in SMC numbers late in the exposure.[189] In addition, thickening of proximal pulmonary arteries in mice exposed to hypoxia is observed and is characterized by adventitial thickening and fibrosis leading to functional increases in the stiffness of these arteries.[190,191] This section of the current review, however, focuses not on hypoxia-induced remodeling, but rather on spontaneous remodeling of pulmonary arterial wall in mice due to alteration in genes implicated in PH pathogenesis.
One example of how a change in the gene dose affects structure and function of the pulmonary artery occurs with a mutation in the BMPR2 gene. Recent reports have demonstrated that BMPR2 signaling plays a critical role in the pathogenesis of both idiopathic and familial PAH. The fact that haploinsufficiency in BMPR2 caused by mutations to the BMPR2 gene correlate with the development of PAH in patients inspired development of mice heterozygous for the BMPR2-allele (BMPR2+/−) in a attempt to create an animal model of the disease.[192,193] One study has reported that BMPR2+/− mice show moderately elevated mean pulmonary artery pressure and a modest increase in pulmonary vascular resistance under basal condition.[193] However, other studies using the same mouse strain did not detect any significant differences in RV systolic pressure between BMPR2+/− and control mice.[194,195] Liu et al. have also found that in mice in which BMPR2 was constitutively knocked down using a short hairpin RNA did not exhibit increases in pulmonary vascular resistance.[196] In contrast, mice with SMC-specific, as well as endothelial-targeted downregulation of BMPR2 signaling using a dominant-negative form of BMPR2 have elevated RV systolic pressure, increased pulmonary artery muscularization, and increased inflammatory indexes.[192,197] West et al. have also recently reported that mice expressing a BMPR2 mutation in the tail domain in SMC develop PH characterized by significant vascular remodeling, pruning, and adventitial perivascular inflammation.[198] In another study, Hong et al. have demonstrated that complete loss of BMPR2 in endothelial cells is sufficient to generate pulmonary vascular remodeling and spontaneous PH.[199] Overall, then, BMPR2 mutation appears to drive elevated pulmonary vascular resistance through multiple mechanisms, but spontaneous occlusion or dropout of vessels seems to be central in at least some variations on this model.[198–200]
Defects in serotonin signaling have long been associated with idiopathic PAH. Although, in normoxia, transgenic serotonin transporter-deficient mice have been shown to present with normal hemodynamics, these mice develop a mild form of PH under exposure to chronic hypoxia.[201] Despite the presence of potentiation of hypoxic pulmonary vasoconstriction in these knockout mice, the exposure to hypoxia also results in vascular remodeling and ventricular hypertrophy greater than that observed in wild-type mice under identical hypoxic conditions. Remodeling is also induced by the increase in serotonin transporter function, as transgenic mice overexpressing the serotonin transporter under the control of the SM22 promoter spontaneously develop PH with marked increases in right ventricular systolic pressure and pulmonary arteriolar remodeling in normoxia.[202] However, these mice under hypoxia have a lesser degree of pulmonary vasoconstriction when compared with wild-type mice-a response that might be explained by the production levels of serotonin. Collectively, the findings from the serotonin transporter studies are consistent with the idea that disruption of the physiological serotonin levels i.e., either increase or decrease in serotonergic tone, provokes structural changes in the vessel wall.
The activity of vasoactive intestinal peptide (VIP) also regulates tone and makeup of pulmonary vasculature, as deletion of VIP leads to moderately severe PAH during air breathing.[203] This model displays in addition to RV hypertrophy and RV hypertension, thickening of the PA, as well as its smaller branches. The emerging importance of the VIP signaling in PAH is underscored by the fact that VIP-related pituitary adenylate cyclase-activating peptide also has VIP-like actions on the pulmonary circulation, which include vasodilation and inhibition of vascular remodeling.[204] Mutant mice lacking the principal receptor for the VIP-related pituitary adenylate cyclase-activating peptide spontaneously develop severe PH and right ventricular failure, resulting in death within the first postnatal weeks.[205] Thus, deletions of either VIP or the main receptor for VIP-related pituitary adenylate cyclase-activating peptide offer additional experimental models of PH useful for studies of spontaneous development of PAH.
While the above two examples described vascular remodeling in models which rely on ligand or its receptor-mediated structural modifications to pulmonary vasculature, there is now growing evidence for the role for enzymes that control protein phosphorylation levels in development of PH. Phosphatase and tensin homolog (PTEN) is a case in point. As a dual-specificity lipid and protein phosphatase, PTEN, inhibits cell proliferation, survival, and growth predominantly through dephosphorylation of phosphatidylinositol 3,4,5-triphosphate, thus antagonizing phosphatidylinositol 3 (PI3)-kinase-mediated signaling events.[206–208] SMC-targeted PTEN-deficient mice spontaneously develop features of pathological vascular remodeling in both the systemic and pulmonary vasculature that are mediated, at least in part, through the induction of the chemokine, stromal cell-derived factor-1α or SDF-1α[209] Natural occurrence of right ventricular hypertrophy in PTEN knockout mice suggests the development of PH. Indeed, PTEN-deficient mice exhibit increased wall thickness of small pulmonary arteries, occluded precapillary arterioles, and reduced alveolarization compared to wild-type mice. PTEN is not the only phosphatase affecting the structure and composition of the pulmonary artery, as MAP kinase phosphatase 1 (MKP1) is also known for such structural effects. MKP-1 deficient mice are susceptible to hypoxia exposure as development of severe hypoxia-induced PH and remodeling of pulmonary arteries were demonstrated in these mice.[210–211] Therefore, SMC-targeted PTEN-deficient mice and MKP1 null mice are very useful in studies of the role of phosphatases in the pathophysiology of PH.
Recently, Maurer et al. reported that fos-related antigen 2 (Fra-2) transgenic mice, which ectopically express Fra-2 in various organs, including lungs, develop severe vascular remodeling of pulmonary arteries resembling human systemic sclerosis-associated PH[212] (Fig. 3). Such pattern of expression was achieved by driving the expression of the murine fra-2 gene with the ubiquitous major histocompatibility complex class I antigen H2Kb promoter.[213–214] These features of the Fra-2 transgenic mouse model of PH are consistent with the observed overexpression of Fra-2 protein in the skin and lungs of patients with systemic sclerosis.[213,214] Histopathological features of Fra-2 transgenic mice are typical for systemic sclerosis-associated PAH, and include the following: Intimal thickening with concentric laminar lesions, medial hypertrophy, perivascular inflammatory infiltrates, and adventitial fibrosis.[212] Principally, myofibroblasts, and to a lesser extent VASMCs, were found to be key players in the remodeling of pulmonary arteries in the Fra-2 transgenic mice. In contrast, pulmonary occlusive venopathy has not been frequently detected in this model. Therefore, the Fra-2 transgenic mice represent a new addition to the models of spontaneous PAH and will be specifically useful in modeling systemic sclerosis-associated PAH.
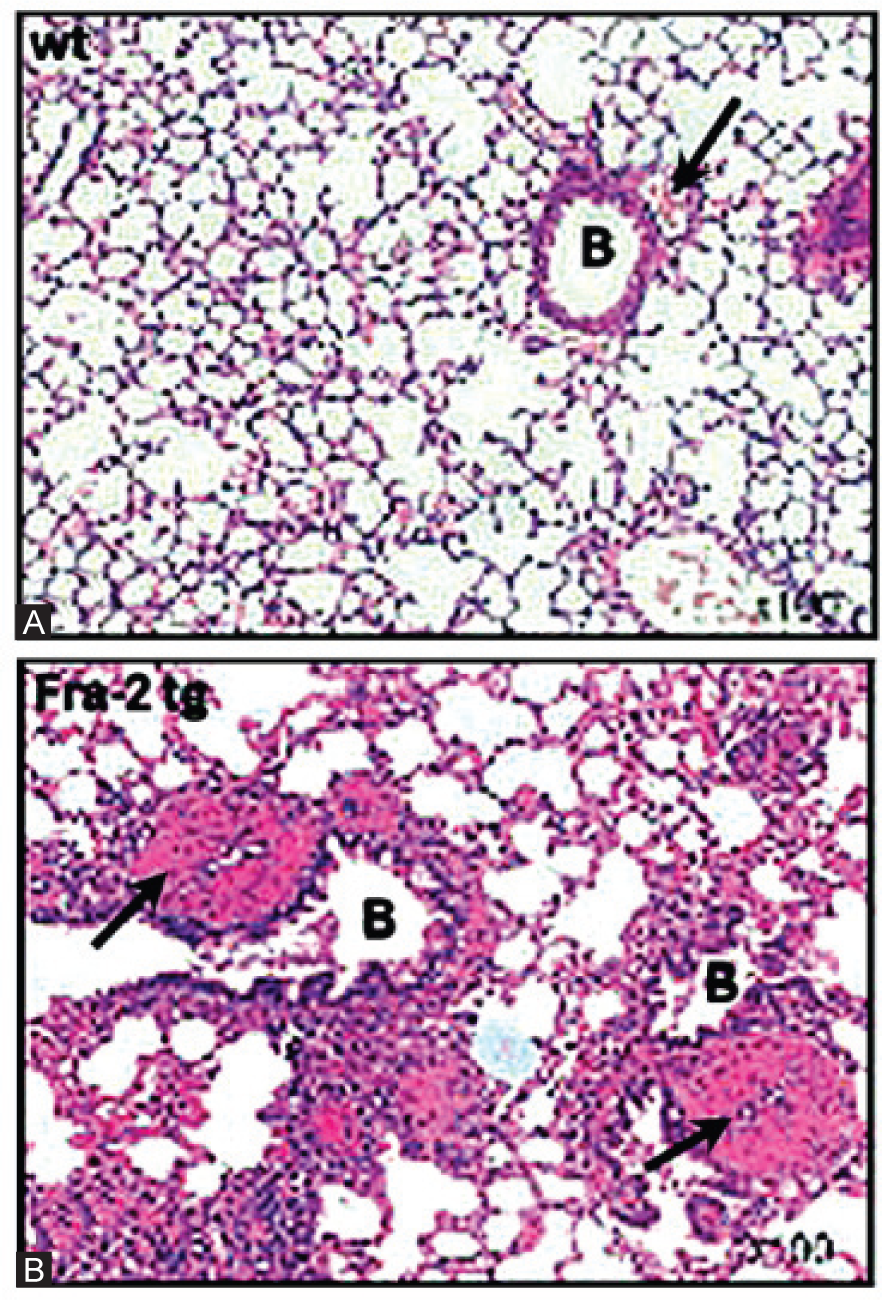
Pulmonary pathology of Fra-2 tg mice. Vascular remodeling
It is important to emphasize that even though the murine models generated to date exhibit high degree of vascular muscularization, elevated pulmonary artery blood pressure can be measured in the absence of significant pulmonary arterial muscularization in mice.[93,192,215,216] A recent report clearly demonstrates that pulmonary arterial muscularization induced by a Th2 immune response failed to cause PH, even when muscularization was advanced and affecting a significant area of the lungs.[217] Clinical observations in pediatric patients with a congenital heart defect associated with remodeling of the pulmonary arteries add further credence to the above-mentioned studies in mice. Commonly, in such patients, the pulmonary arterial pressure normalizes the day after surgical heart repair despite the continuing presence of pulmonary arterial muscularization.[218,219]
In conclusion, PH is a complex disease propagated by multifactorial pathological drivers and events. Although no single murine model of PH generated to date replicates all salient features of the human disease, these models offer by the way of genetic modification of specific genes of interest and consequent distinctive pathological states critical insights into the mechanisms and pathophysiology of PH. Future research progress in this area will undoubtedly help unfold the mystery behind the structural remodeling of pulmonary arteries observed in patients suffering from PH, and by doing so, lead the way toward development of therapeutic strategies that prevent disease progression or approaches capable of reversing the structural abnormalities in the pulmonary vasculature.
INFLAMMATORY MODELS OF PULMONARY HYPERTENSION
Lung inflammation has been implicated as one of the potential mechanisms underlying the development of PH. Infiltrates of leukocytes, as well as expression of various inflammatory cytokines and chemokines including interleukin (IL)-1β, IL-6, and monocyte chemoattractant protein-1 (MCP-1) are increased in the pulmonary arterial wall of PH patients.[220–224]
The ability of inflammation to directly induce PH has been demonstrated in animal models in which chronic inflammation alone was shown to promote pulmonary vascular remodeling and lead to elevation in pulmonary artery pressure.[217,225–228] Hypoxia, a well-known inducer of PH in experimental animal models, also stimulates an inflammatory response.[229–231] In the process of doing so, hypoxia induces an increase in the expression of adhesion molecules such as intracellular adhesion molecule-1 and vascular cell adhesion molecule-1, as well as cytokine MCP-1, all of which support the recruitment of monocytes to the endothelium and thereby promote inflammation in the lung.[224,232]
Although, the levels of inflammatory cytokines are increased in PH patients, as well as in experimental model of PH, the specific role of each cytokine in the pathobiology of PH remains unclear. Recently, the generation of transgenic mice using either overexpression or elimination strategies modulating proinflammatory cytokine expression is providing some crucial mechanistic insights about specific cytokines and their potential roles in the disease process. One such example is circulating IL-6, which is increased in most forms of human PH.[221,233,235] Several experimental studies in mice have also reported an active role for IL-6 in pulmonary vascular remodeling and hypoxic PH.[229] To appraise further the increased expression of IL-6 in PH, Steiner and colleagues have created transgenic mice with lung-specific overexpression of IL-6[228] and Savale et al. have characterized IL-6 knockout mice.[236] Both studies reported a correlation between IL-6 expression and the development of vasculopathy, reproducing conditions characteristic of severe PH in humans. Despite this progress, the mechanism by which IL-6 may contribute to vascular remodeling is not well understood. Since, hypoxic IL-6 null mice have blunted inflammatory cell recruitment, as well as vessel wall remodeling in the lungs, IL-6 might affect pulmonary vascular remodeling by directly targeting vessel wall cells or by indirect effects mediated by inflammatory cells themselves.
Another important cytokine to discuss in the context of inflammatory processes in the lung and their regulation is adiponectin (APN)-an anti-inflammatory adipokine (aka adipocytokine). APN has been implicated as a regulator limiting lung inflammation in PH and thereby reducing the severity of the disease.[101] Adramatic increase in pulmonary arterial muscularization and PH has been observed in the APN null mice relative to wild-type mice in the setting of allergic vascular inflammation.[237] Furthermore, mice deficient in APN develop spontaneous age-dependent increases in accumulation of perivascular inflammatory cells and elevated pulmonary artery pressure suggesting a primary role for APN in vascular inflammation and remodeling. APN null mice also acquire an exaggerated eosinophilic vascular response to allergic lung inflammation.[237] Interestingly, elimination of eosinophils in these APN-deficient mice prevents development of PH.[238] Although, the molecular mechanisms for the anti-inflammatory activity of APN have not been well-defined, findings to date suggest that APN might act directly on proinflammatory cells.
Proinflammatory cytokines and adhesion molecules are not the only mediators of inflammatory processes in the lung. Increased inflammation has also been reported in transgenic mice overexpressing S100A4 (Mts1, a tumor-associated calcium binding protein) in the lungs.[239] Immunoreactivity for S100A4 protein has been localized to the pulmonary artery endothelial cells of mice that developed abnormal plexiform-like lesions in association with an increased number of inflammatory cells. This peri-arterial inflammation is not observed in normal mice nor has it been detected in transgenic, non-diseased mice. The presence of numerous inflammatory cells surrounding affected vessels in diseased transgenic mice suggests that an inflammatory or infectious insult triggers cellular events that lead to the development of plexogenic arteriopathy in these transgenic mice.
In addition to the mechanisms described above that affect inflammation in PH, the fibrinolytic system has been also implicated to play a role in this pathogenic process.[240] For example, activated thrombin-activatable fibrinolysis inhibitor (TAFI), a circulating plasma glycoprotein, inhibits fibrinolysis and therefore its absence would be expected to increase fibrinolysis and ameliorate PH. Indeed, mice deficient in TAFI are protected from monocrotaline-induced increases in vascular remodeling and PH. Further supporting this idea is a finding that induction of PH with monocrotaline is associated with more lung inflammation in wild-type mice than in TAFI-deficient mice.[241]The exaggerated inflammatory response in wild-type mice compared to TAFI-deficient mice may be explained by high concentrations of the inflammatory mediators such as TNFα, IL-6, and MCP-1 in bronchoalveolar lavage from the wild-type mice.
The body's natural pulmonary vasodilator, vasoactive intestinal peptide (VIP), might play a protective role limiting pathogenic mediators in PH, such as those that drive inflammation. One study reports that VIP is absent in pulmonary arteries from patients with idiopathic PAH.[242] Clusters of inflammatory cells, predominantly mononuclear infiltrates, have been observed in the lungs of mice lacking VIP, suggesting that in addition to VIP's role in promoting vasodilation and inhibition of vascular remodeling, VIP engenders an anti-inflammatory environment.[203]
Mutations in the BMPR2 gene have been identified in PAH patients. However, low disease penetrance due to loss of function of BMPR2 gene suggests that additional factors might play a role in the pathogenesis of PH. Indeed, exposure to inflammatory stressors enhanced the severity of PH in BMPR2+/− mice.[195] However, a recent report by Mushaben et al. suggests that vascular remodeling and PAH resulting from chronic allergic inflammation occur independently from the BMPR2 pathway.[243] The discrepancies between these two studies may be due to the induction of different types of inflammatory responses.
Recently, Hara et al. identified a trans-membrane protein called multidrug resistance-associated protein 4 (MRP4) as an important regulator of PH and present evidence from patients, as well as from mice supporting a proinflammatory role for the MRP4.[244] Two lines of evidence in support of this idea are as follows: (1) That MRP4 is overexpressed in human lungs from patients with PAH; and (2) that MRP4 deficient mice are resistant to the development of hypoxia-induced PH.[244] This function for MRP4 in PH is logical since MRP4 is a membrane transporter for prostaglandins-an anti-inflammatory prostaglandin E1 and a proinflammatory prostaglandin E2.[245] In addition, this study demonstrates that MK571, an MRP4 inhibitor, reverses the progression of hypoxia-induced PH in wild-type mice. Reduction in vascular remodeling observed after MRP4 inhibition is associated with reduction in the release of proinflammatory factors from activated endothelial cells and smooth muscle cells. However, there is no evidence that MRP4 directly controls the release of such factors or directly modulates the function of inflammatory cells even though MRP4 expression has been reported in blood cell progenitors.[245] Whether MRP4 prevents the development of PH through a direct effect on the inflammatory process, in addition to its action on vascular endothelial and SMCs deserves further investigation. And following this line of reasoning, it would be beneficial to investigate inhibition of MRP4 as a potential therapy in human PAH.
Together these studies provide credence to the hypothesis that inflammation is an important component of the PH pathogenesis and suggest that activation of anti-inflammatory pathways could play an important therapeutic role in treating this disease. Indeed, several inflammation suppressing treatments, including the use of an IL-1-receptor antagonist and glucocorticoids,[240,247–253] have been shown to prevent experimentally-induced PH when used early in the course of monocrotaline-induced PH in rats. Glucocorticoids have also been shown to reverse established PH and improve survival during later disease stages in this model.[254] Of several potential glucocorticoid-mediated mechanisms, a reduction in IL-6-expressing adventitial inflammatory cells, as well as a direct antiproliferative effect on smooth muscle cells, have been observed.[254] The ongoing challenge is to fully characterize the interplay between pro and anti-inflammatory processes occurring in PH and, in that context, to determine whether therapeutic strategies aiming at suppressing inflammation will be of therapeutic benefit to patients with PH.
CONCLUSIONS
A central message of this review is that transgenic mouse models have unparalleled power for unraveling the mechanisms behind specific processes, including regulation of pulmonary vascular tone, energy metabolism, remodeling, and inflammation. Each of these processes is involved in the development of human pulmonary hypertension, and some of them are probably etiologic for specific WHO classifications of human PH. Many of the mouse models discussed here are not widely known in the PH community, but have contributed extensively to our knowledge of how tone is regulated, how the pulmonary vasculature responds to injury, which pathways are likely involved in adaptive versus maladaptive inflammation and repair, and how regulation of energy metabolism underlies many of these processes.
One of the primary difficulties that the field has faced has been poor “bench to bedside” translation, from rodent models to human therapies. There are two key reasons for this. First, the most popular rodent models are popular because they're easy, not because they accurately model the molecular etiology of any human disease. Very few humans have developed PH as a result of ingesting toxic plant alkaloids, and while hypoxia is etiologically relevant to some classes of human PH, it's generally not relevant to the classes it's most used for. Second, even in those mouse models which do share molecular etiology with a human condition, such as with BMPR2 mutations, mice do not follow the same course of development as the human disease. The time for development of disease is greatly compressed, and the pulmonary physiology of mice is substantially distinct from human disease.
The first of these problems can be overcome by increasing the care with which we use models. The second problem, however, is fundamental. We can use mice to model molecular events, and specific physiologic processes, with great fidelity to the human condition. We can not use them to model the natural history of any disease.
The power of transgenic mouse models, then, is in modeling process, and excellent models exist for most of the processes thought to be important in the development of many of the different classes of PH. In the best case, drugs found to effectively intervene against molecular defects in mice will be effective in correcting the same molecular defect in human patients; we are just coming into an era of new therapies designed in this way. This space, between the simplicity of cell culture and the complexity of human trials, is the space occupied by animal models, and in which they are most powerful.