
Abstract
Although medical therapies for pulmonary arterial hypertension have greatly improved, it remains a chronic and fatal disease. For patients who are refractory to medical therapy, lung transplantation is an important treatment option. This review discusses issues pertaining to indications for transplant, preparation for transplant and listing, operative issues, and outcomes for patients with pulmonary arterial hypertension.
INTRODUCTION
Pulmonary arterial hypertension (PAH), initially described hemodynamically in 1951 by David Dresdale, was a disease that for decades had no effective treatment.[1] Prior to the transplant era, the chances for survival with pulmonary hypertension were grim, with median survival only 2.8 years.[2] However, two early discoveries changed the prognosis for patients with pulmonary arterial hypertension: lung transplantation and the discovery of epoprostenol. Heart-lung transplantation developed as a surgical means to treat pulmonary vascular disease, and the first heart-lung transplant, was performed at Stanford by Norman Shumway, John Wallwork, and Bruce Reitz in 1981.[3] Soon after the first successful heart-lung transplants, single and bilateral lung transplantation evolved.[4–6] Joel Cooper reported the first single lung transplant in a patient with pulmonary fibrosis in 1986, and soon the bilateral sequential lung transplant technique became favored.[4–6] With early surgical successes, the number of lung transplants registered per year worldwide continues to grow, surpassing 2700 transplants in 2009 (Fig. 1).[7]
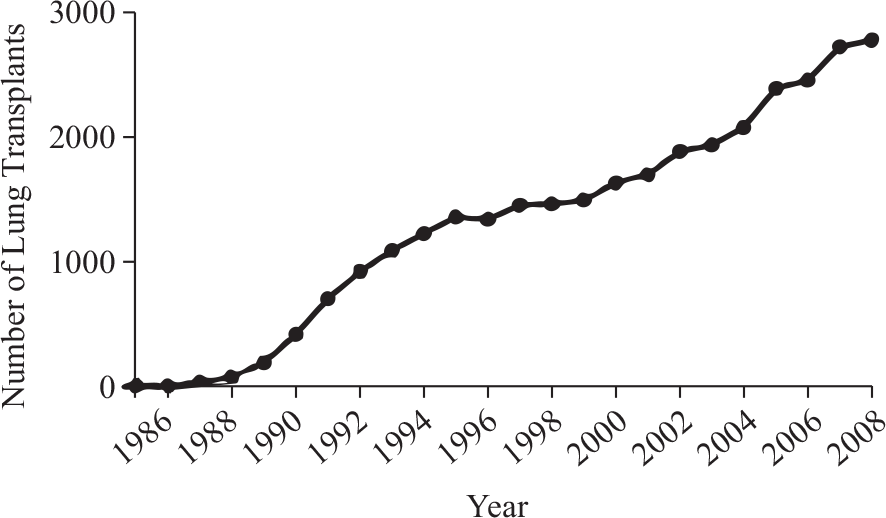
Since the first lung transplant in 1986, the number of lung transplants continues to grow.[7]
This early success in surgical treatment of PAH was soon followed by the major medical discovery of prostacyclin (epoprostenol)[8,9] A landmark randomized control trial demonstrated that intravenous epoprostenol produced improved symptoms, hemodynamics, and increased survival in patients with primary (idiopathic) PAH, thereby changing the course of this disease.[10,11] In a randomized controlled trial, epoprostenol was also associated with a 5-year survival of 55%.[12] Since this medical breakthrough, there have been many more discoveries, with the development of new prostacyclin analogs (treprostinil, iloprost) as well as development of other classes of medications.[13–16] These include endothelin receptor antagonists (bosentan, ambri-sentan)[17,18] phosphodiesterase-5 (PDE5) inhibitors (sildenafil, tadalafil),[19] and newer agents currently under study (imatinib, riociguat).[20,21] These medications have been shown to improve hemodynamics, symptoms, exercise capacity, time to clinical worsening, and quality of life in patients with idiopathic PAH. A recent meta-analysis combined clinical trials of goal-oriented PAH therapy, and showed that active treatment was associated with an overall reduction in mortality of 43% (P=0.048).[22]
The past 30 years have been notable for parallel developments in surgical and medical therapies for pulmonary hypertension (Fig. 2). Although successful developments in the medical treatment of patients with PAH has decreased the number of patients listed for lung transplantation, there remain patients who are refractory to, or progress despite multidrug therapy. For this population, lung transplantation often remains the only viable option for improving survival. This review article will address the indications for lung transplant in PAH, specific operative considerations unique to patients with PAH, and transplant outcomes in patients with PAH, both in terms of postoperative physiologic changes and survival.
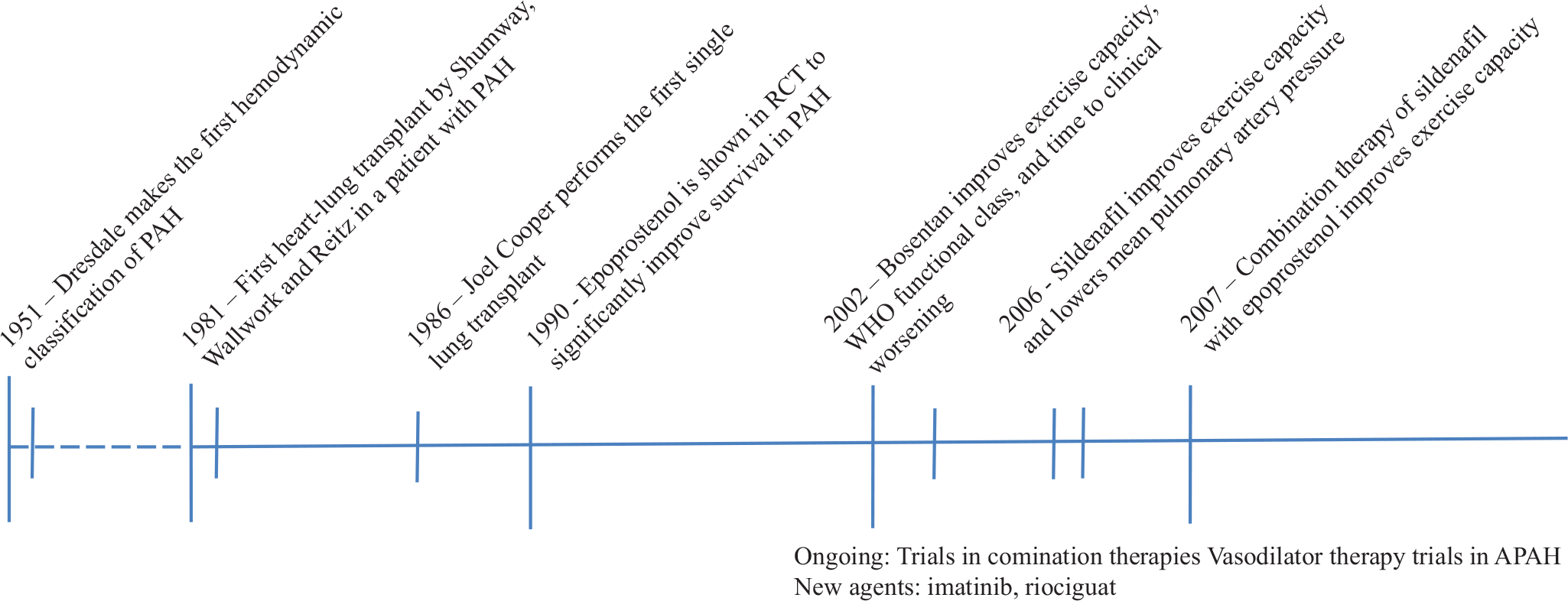
Timeline of major medical and surgical developments in the treatment of pulmonary arterial hypertension.
Definitions and nomenclature in PAH
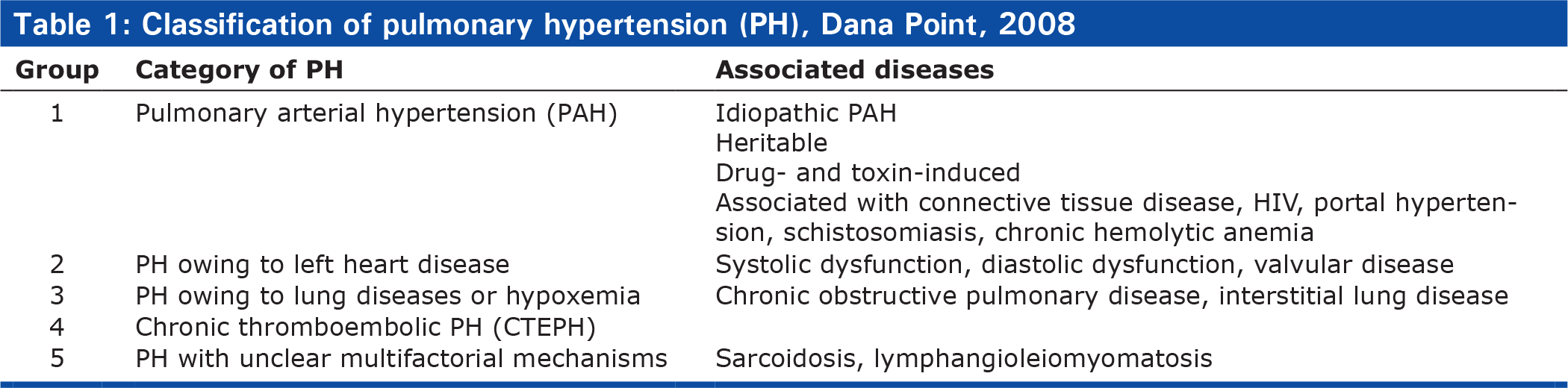
PAH is defined by a mean pulmonary artery pressure greater than 25 mmHg at rest and a normal pulmonary capillary wedge pressure of 15 mmHg or less with a pulmonary vascular resistance greater than 3 Wood units.[23] As treatments have developed for PAH, so has our understanding and classification system. The initial classification scheme in 1973 involved only two categories: primary and secondary PAH.[1, 24] This was expanded into 5 major categories at the Second World Symposium on PAH in Evian, France in 1998. Categorization into groups based on putative similar pathophysiology helped investigators to conduct clinical trials, and led to the approval of 8 medicines for PAH.[24] In 2003 at the Third World Symposium on PAH in Venice, Italy, the terms primary and secondary PAH were eliminated, although some people still use these terms today.[25] Finally in 2008 the Fourth World Symposium of PAH at Dana Point, California, the nomenclature was further revised to better group similar diseases and account for mutations, and continues to be the nomenclature in use today (Table 1).[24] Lung transplantation has been performed commonly for patients in diagnostic groups 1,3, and 5. Patients with idiopathic pulmonary arterial hypertension (IPAH) comprise 3.3% of all transplant recipients and 5.4% of double lung transplant recipients.[7] This article will pertain mainly to patients with IPAH unless otherwise stated.
Classification of pulmonary hypertension (PH), Dana Point, 2008
INDICATIONS: REFERRAL FOR LUNG TRANSPLANT IN PAH
Although we have made considerable progress in the medical management of PAH, not all patients respond equally well to medications. In addition, those who respond initially may suffer a sudden decline in clinical status. It is recommended that lung transplantation be utilized for patients who do not respond to optimal vasodilator therapy (Table 2). Sitbon and colleagues identified a high-risk subset of patients as those who 3 months following initiation of prostacyclin therapy remain in New York Heart Association functional class III or IV or do not achieve a 30% drop in pulmonary vascular resistance from baseline.[12] Other risk factors associated with poor outcomes include hyponatremia, hyperbilirubinemia, echocardiographic evidence of severe right ventricular dysfunction measured by tricuspid annular plane systolic excursion (TAPSE) less than 1.8 cm, and six minute walk distance less than 332 m.[26–29]
Guidelines for referral for lung transplantation

Although much progress has been made in treatment of idiopathic pulmonary hypertension (IPAH), additional risk factors to consider in patients with PAH include history of underlying connective tissue disease (CTD) or other primary lung disease. In a study of patients in the REVEAL registry, patients with scleroderma had higher plasma brain natriuretic peptide levels, lower diffusing capacity for carbon monoxide, and significantly worse one-year survival than patients with IPAH (82% in patients with scleroderma vs 86% in all CTD-PAH vs 93% in IPAH).[30] Patients with pulmonary hypertension associated with underlying lung diseases such as chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, and sarcoidosis, also are at increased risk of mortality,[31–38] and those with combined emphysema, interstitial lung disease and pulmonary hypertension have a particularly high mortality risk, with only a 60% one-year survival.[39] Studies have shown variable effects of oral vasodilator therapy in the treatment of PAH associated with lung diseases, presumably due to worsening ventilation/perfusion mismatch.[40–45] This has been somewhat overcome through the use of inhaled prostacyclins, which may have promise in COPD and interstitial lung diseases.[46,47] Nonetheless, responses to such medications in patients with underlying hypoxemia due to parenchymal lung disease are variable, and the potential for worsening persists. Hence in patients with primary parenchymal disease or CTD, the presence of PAH may indicate a need for expedited transplant evaluation.
Given the difficulty in predicting course of illness in patients with PAH, whether idiopathic or associated with pulmonary parenchymal disease (APAH), it is recommended that patients be referred early to a transplant center for evaluation. Early evaluation allows time for physicians and patients to explore any possible risk factors and devise strategies to overcome them (e.g., obesity and weight loss, deconditioning and pulmonary rehabilitation). There are also circumstances in which patients will not be considered viable candidates for lung transplantation. Contraindications include active cancer, substance abuse, noncompliance with medications, and lack of social support. The consensus statement on lung transplantation candidacy was last published 5 years ago,[48] and many would argue that, given our advancements in surgical and medical transplant care, previous contraindications may no longer apply. In addition, criteria for lung transplant candidacy varies significantly among centers, so early evaluation provides an opportunity for opinions at other more aggressive centers if patients are declined at one center. Acknowledging this evolution in practice, it is best to refer patients to the transplant center for preliminary evaluation. In addition, even when patients with PAH are initially too well to be listed for transplant, early evaluation allows for close follow up should the patient's clinical condition decline. Typically patients on the waiting list are seen every three to six months for a medical update and to update their LAS. This is especially important in pulmonary hypertension patients as any change in clinical status may place them at need for LAS appeal and expedited transplantation. Finally, if patients are followed at a tertiary referral center, they may benefit from opportunities to enroll in clinical trials for IPAH or APAH.
PREPARATION: EVALUATION, LISTING FOR TRANSPLANTATION, AND IMPACT OF THE LUNG ALLOCATION SCORE
Full lung transplant evaluation involves a detailed process to best determine the nature and trajectory of a potential candidate's illness, as well as identify possible risk factors for potential modification. Depending on the center, much of the testing may be done locally, if patients and referring physicians prefer, however visit to the transplant center is usually a several day process. In addition to consultations with transplant pulmonology, transplant surgery, and social services, patients also undergo psychiatric screening and participate in a transplant orientation class. Patients also undergo full laboratory, electrocardiographic, radiologic testing, and cardiac catheterization. In addition, they also must be up to date on vaccines and age-appropriate cancer screening (Table 3).
Pre-transplant evaluation includes consultations and visits to evaluate the candidate's pulmonary disease and risk factors for lung transplantation

In 2005, the method in which patients were prioritized for lung transplant underwent a major revision with the development of the lung allocation score (LAS). Prior to this, patients were prioritized on the waiting list by how much time they had accrued on the waiting list. This resulted in many deaths on the waiting list, and compelled physicians to list patients long before they were sick enough to justify the risks of lung transplant. In this era of long waitlist times, when patients' with PAH deteriorated quickly, techniques such as balloon atrial septostomy were utilized by some physicians as a bridge to transplant.[49]
To simplify the listing process, reduce time (and deaths) on the waiting list, and help assure that organs would go to those who were in most need, the LAS was developed.[50] The LAS is a scoring system developed from demographic and clinical characteristics, which takes into account waiting list urgency and the probability of post-transplant survival. The score is normalized to range between 0 and 100, and the sicker a patient is, the higher their score. Patients are placed on the waiting list according to blood type and size, with highest priority going to patients with the highest LAS.
After development of the LAS, the likelihood of transplantation from the waiting list increased for all diagnoses. Deaths on the waiting list decreased for all diagnostic groups except IPAH, which remained unchanged. Under the LAS system, patients with IPAH were less likely to be transplanted than patients with IPF (hazard ratio [HR] 0.53; P<0.001) or CF (HR 0.49; P<0.001) and more likely to die on the waiting list than patients with COPD (HR 3.09; P<0.001) or CF (HR 1.83, P=0.025).[48, 51] Critics of the LAS system state that the factors accounted for in the equations are more heavily weighted towards pulmonary function tests, which do not correlate with disease severity in IPAH. In an analysis comparing mortality predicted by the LAS system to actual mortality in the REVEAL cohort containing 2967 patients with PAH, two additional variables were independently associated with increased mortality compared to the LAS in multivariable analysis: mean right atrial pressure greater than or equal to 14 mmHg and six minute walk distance less than or equal to 300 m.[52] Although modification of the LAS system is under discussion, the United Network for Organ Sharing (UNOS)/Organ Procurement and Transplant Network (OPTN) Thoracic Organ Transplantation Committee has adopted criteria for appeal of an LAS in a patient with IPAH (Table 4). A patient with IPAH will be granted a LAS in the 90th percentile of all lung allocation scores when they satisfy all of the following criteria: the patient is deteriorating on optimal medical therapy, right atrial pressure is greater than 15 mmHg, and cardiac index is less than 1.8 L/min/m2.[53]
Criteria for LAS appeal in lung transplant candidates with IPAH

The impact of the LAS on patients with IPAH on the waiting list highlights the importance of close follow up while patients are on the waiting list, as they may meet criteria for LAS appeal either at time of listing of later in the course of their disease.
OPERATIVE ISSUES: SINGLE VERSUS DOUBLE LUNG TRANSPLANT
Most transplant centers now favor double lung transplantation over single lung transplantation (Table 5). Although lung transplantation began with heart-lung transplantation, with technical improvements in the 1990s, single lung transplantation became the procedure of choice for lung transplant when the left ventricular function was intact. Early studies showed that single lung transplantation is quite effective in lowering pulmonary artery pressures and improving right ventricular function.
Comparison of single, double, and heart lung transplantation

Early experience at the University of Pittsburgh explored outcomes in single versus double lung transplantation for IPAH. In a retrospective study comparing 37 double lung transplant recipients and 21 single lung transplant recipients transplanted between 1989–1996, one-month, one-year, and four-year survival was comparable between groups. As expected, time on cardiopulmonary bypass was significantly shorter in the single lung recipients. Mean pulmonary artery pressures were significantly lower in the double lung transplant recipients at 1 hour, 12 hours, and 24 hours post-transplant (P<0.02). While there was a slightly lower incidence of postoperative diffuse alveolar damage in the single lung recipients (43% versus 51% in double lung recipients), there was a trend toward slightly higher incidence of obliterative bronchiolitis among single lung transplant recipients (9/21, 42.9%) than double lung transplant recipients (9/37, 24.3%, P=0.14).[54] In an earlier study, Bando and colleagues noted less improvement in pulmonary artery pressures and increased graft-related mortality in single-lung transplant recipients compared with double lung and heart-lung recipients.[55] In another study comparing single lung transplant recipients with and without pulmonary hypertension, Bando and colleagues found that pre-operative pulmonary hypertension was associated with significantly lower one-year survival (53% versus 72%; P<0.05) and New York Heart Association functional class (P<0.05).[56]
At Johns Hopkins, Conte and colleagues reviewed the outcomes of all single and double lung transplants performed for IPAH or PAH associated with CTD or primary lung disease. In their review, patients with primary (idiopathic) PAH who received a double lung transplant had better survival than those who received a single lung transplant.[57] Among patients with CTD-associated PAH and APAH (secondary PAH), there was a trend towards improved survival in double lung transplant when the mean pulmonary artery pressure was greater than 40 mmHg.[57] Although single lung transplantation is feasible,[58–60] based on these findings and clinical experience, the majority of centers now favor double lung transplantation for PAH.
Improvement in surgical techniques with double lung transplantation as well as the ability of the right ventricle to recover postoperatively have rendered heart-lung transplantation much less common. In fact, most centers try to avoid heart-lung transplantation when possible, due to increased mortality as well as scarcity of organs. In the most recent analysis of the International Society of Heart Lung Transplantation (ISHLT) registry, median survival in heart-lung transplantation for IPAH was 3.8 years.[7] However, when a patient's disease progresses to the point to right ventricular failure requiring inotropic support, they may require heart-lung transplantation rather than double lung alone. Right ventricular function, therefore, helps determine the window for lung transplantation in PAH.
OUTCOMES: PHYSIOLOGIC IMPROVEMENTS AND SURVIVAL POST LUNG TRANSPLANT
Immediate peri- and post-operative care in patients transplanted for PAH requires close monitoring and often may require temporary inotropic, vasopressor, and inhaled nitric oxide support. While patients no longer require their pulmonary hypertensive medications, they may require some support of the right ventricle as it recovers. In addition, patients should be monitored closely for the development of primary graft dysfunction in the first 72 hours. Given the complexities of peri-operative management in patients transplanted for PAH, it is recommended that they are managed at a higher volume center with experience in this disease.
The response of the right ventricle to lung transplantation is immediate and remarkable (Fig. 3). Patients are often admitted for surgery on combination vasodilator therapy, only to be completely off pulmonary hypertensive medications with dramatic improvement in pulmonary artery pressures. These improvements are evident in the operating room, as described in a prospective study of intraoperative TEE data: the mean pulmonary artery pressure in those with severe pulmonary hypertension decreased from 76±14 mmHg to 31±11 mmHg (P<0.05) immediately after lung transplantation.[61] In another study looking at hemodynamic data in seven single lung transplant recipients just beyond the immediate postoperative period into the early postoperative period (mean 13 weeks post transplant), mean pulmonary artery pressures decreased from 64±18 mmHg to 18±5 mmHg (P=0.001), and pulmonary vascular resistance index decreased from 1924±663 dyne sec cm-9 to 233±73 dyne sec cm-9 (P=0.001).[60] In a retrospective review of 100 consecutive patients transplanted for idiopathic pulmonary hypertension or pulmonary hypertension secondary to congenital heart disease, reductions in mean pulmonary artery pressures (from 65.9±13.1 mmHg pre-transplant to 21.9±5.9 mmHg (P<0.001) and pulmonary vascular resistance (18.8±8.0 Woods Units pre-transplant to 2.1±0.9 Woods Units post-transplant, P<0.001) were seen at 24 hours and sustained at 1 year later, and improvement in right ventricular ejection fraction was notable at one year (26.8±12.6% pre-transplant to 56.6±8.8% at 1 year, P<0.001).[62] These results were seen in both single and double lung transplant recipients.
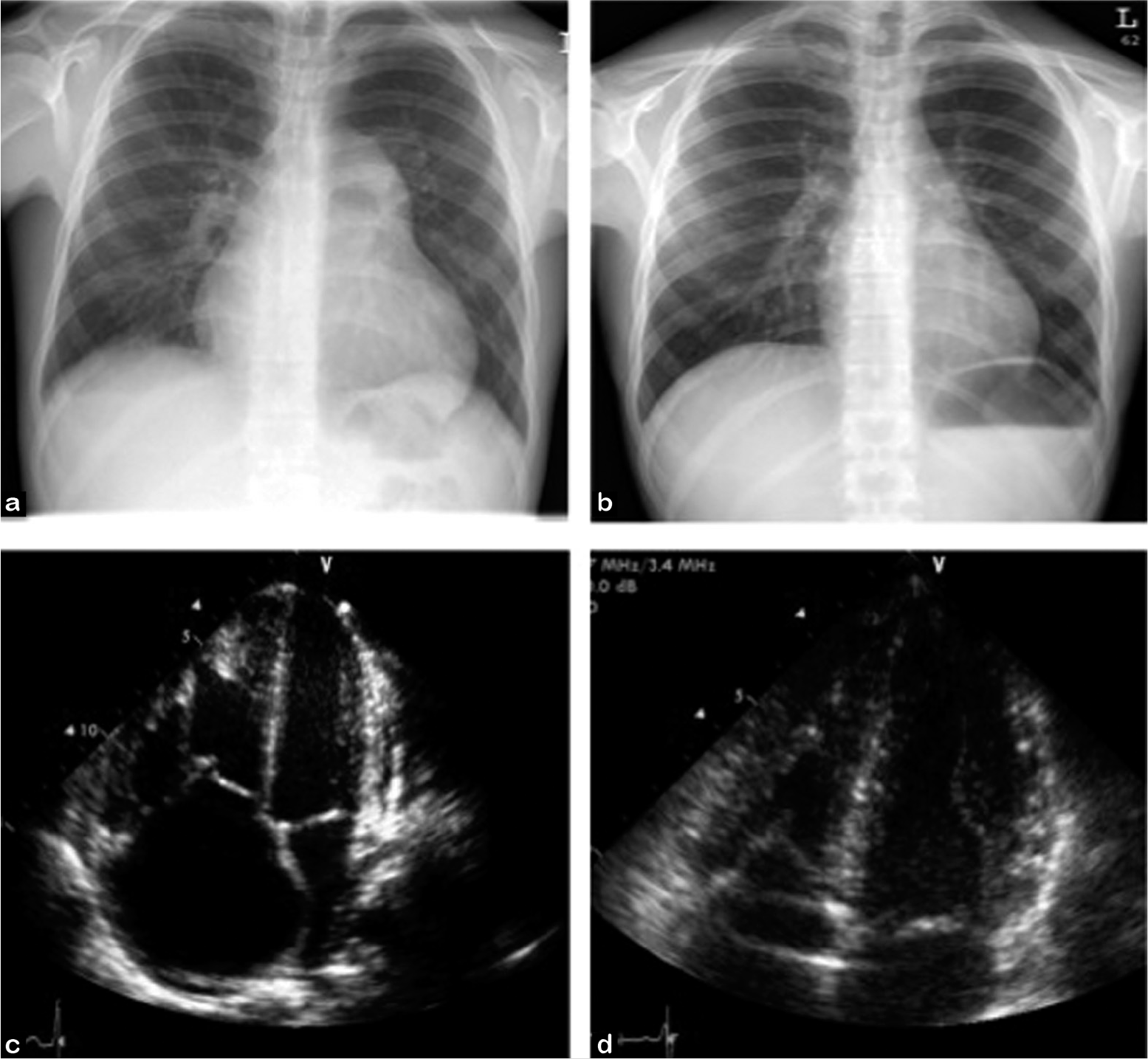
Double lung transplantation is an effective treatment for PAH. Chest radiographs (a) pre- and (b) post-double lung transplant demonstrate radiographic resolution of enlarged pulmonary arteries and right ventricular remodeling post transplant. Echocardiographic images (c) pre- and (d) post-transplant demonstrate resolution of right atrial enlargement and septal bowing seen in severe PAH.
Lung transplantation provides an opportunity for patients to extend their lives as well as improve quality of life. According to the most recent ISHLT registry data, overall median survival for all lung transplant recipients transplanted between January 1994 and June 2008 was 5.3 years, and among those who survive at least 1 year, median survival was 7.5 years.[7] When analyzed by eras in transplantation, overall median survival has significantly improved. Patients transplanted from 2000 through June 2007 have a median survival of 5.7 years, compared with 4.7 and 4.2 years in patients transplanted from 1998 to 1994, and 1995 to 1999, respectively.[7]
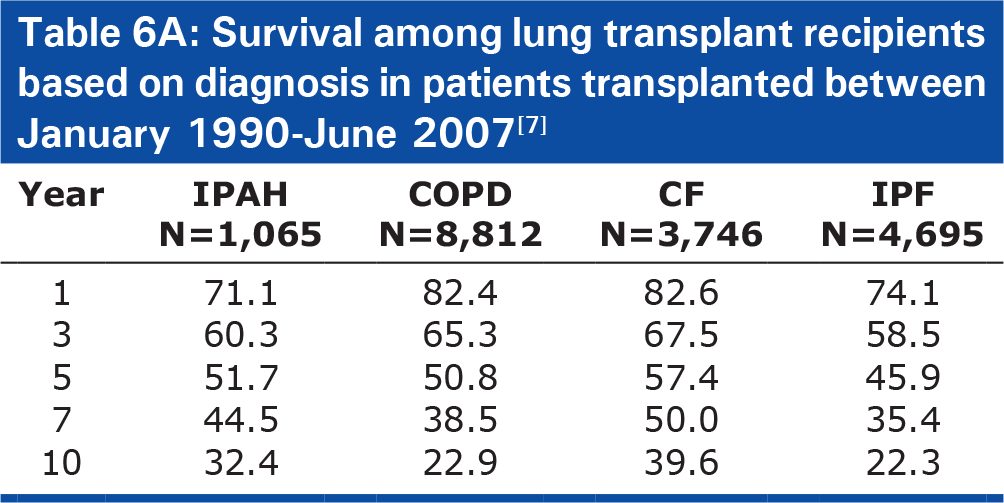
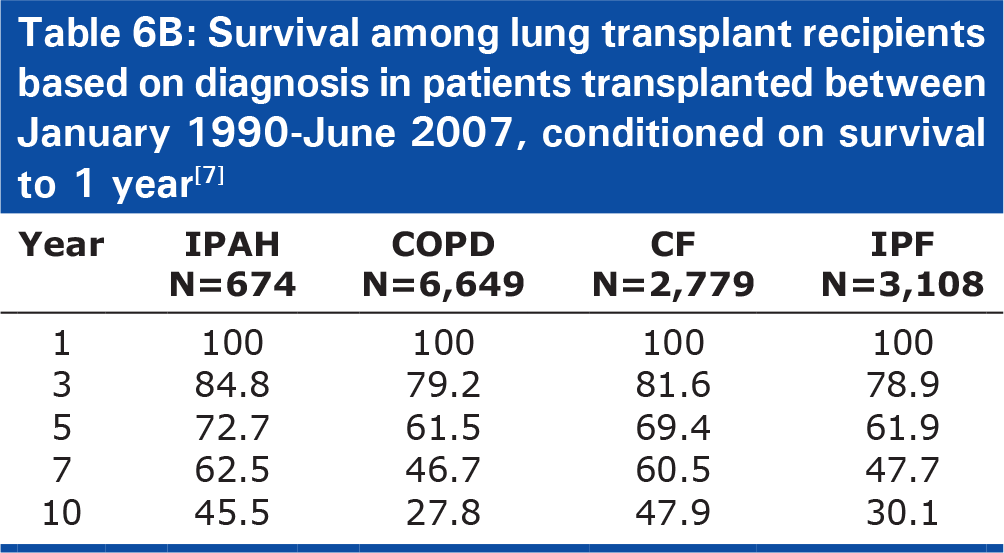
Patients with IPAH have a greater short-term risk after transplant, but also a better chance of long term survival. Patients with PAH have the lowest 3-month and 1-year survival rate (76% and 71.1%, respectively), when compared to patients with IPF (85%, 74.1%), CF (90%, 82.6%), and COPD (91%, 82.4%), see Table 6A).[7] In fact, in multivariable analysis, a diagnosis of IPAH was the greatest categorical risk factor for one-year mortality (relative risk [RR] 2.19, 95% confidence interval [95%CI] 1.63–2.95, P<0.0001).[7] However, when lung transplant recipients with IPAH live for at least one year, they have improved long-term survival compared to patients with other underlying lung diseases. Accounting for risk of early mortality by conditioning survival on living one year, patients with IPAH had a lower risk of 5-year mortality (RR 0.51, P=0.0032) and significantly improved long-term survival (median survival 9.3 years) compared with patients with COPD (6.6 years, P=0.01) and IPF (6.7 years, P<0.0001), Table 6B).[7]
Survival among lung transplant recipients based on diagnosis in patients transplanted between January 1990–June 2007[7]
Survival among lung transplant recipients based on diagnosis in patients transplanted between January 1990–June 2007, conditioned on survival to 1 year[7]
In lung transplant recipients who die within the first year, the most common causes of death are non-CMV infections and graft failure.[7] Within the early post-operative period, primary graft dysfunction (PGD) is a significant concern in patients with IPAH (as well as in other patients with increased pulmonary arterial pressures in general), and is associated with increased risk of death.[63,64] PGD is defined as lung injury occurring within the first 72 hours post-transplant, and is considered grade 2–3 when the PaO2/FiO2 ratio is less than 300 and bilateral infiltrates are present on chest radiograph.[65] While the exact pathogenesis is unknown, pulmonary hypertension has been repeatedly associated with increased risk of PGD, and researchers hypothesize that increased shear stress associated with elevated pulmonary artery pressures during reperfusion may play a role.[64, 66–68] Several studies have identified a diagnosis of IPAH as a risk factor for developing PGD. Others have identified elevated pulmonary artery pressures as a risk factor for developing PGD in non-IPAH patients.[68,69] The difficulty with many of these studies, however, is that IPAH and pulmonary artery pressures cannot be completely isolated from other risk factors for PGD, namely use of cardiopulmonary bypass and blood products.
While the ISHLT data seem to indicate that the diagnosis of IPAH is a risk factor for one-year survival post transplant, it is quite possible that at high-volume centers that specialize in transplant for IPAH, survival is better than ISHLT data. A recent study showed that in general, high-volume transplant centers are associated overall improved five-year survival.[70] In a retrospective study of patients with PAH at the University of Pittsburgh, which has performed over 60 lung transplants per year over the past 7 years, one-year survival in cohort of 30 patients transplanted between 1994–2006 was 86%, compared to 66% in the comparable ISHLT cohort, and 58% in IPAH patients transplanted at the University of Pittsburgh from 1982 to 1993.[71] The authors attributed their improved outcomes to improvements in donor and recipient surgical techniques as well as improvements in postoperative medical management of transplant recipients[71] Similarly, a retrospective study of 220 patients transplanted between 1986 and 2008 at Marie-Lannelongue Hospital in Paris reported a 79% one-year survival among double lung transplant recipients in patients with IPAH.[72] These studies suggest potential benefit to performing transplants for PAH at experienced high-volume centers.
Most lung transplant recipients enjoy improved functional status post transplantation. In a single center survey comparing 54 lung transplant recipients to 44 transplant candidates, recipients reported higher levels of happiness with their life and health as well as improved functional status.[73] In a cross-sectional analysis of the functional status of lung transplant recipients in the ISHLT dataset, over 80% of survivors reported no limitations in activities, and less than 3% reported need for full assistance with activities.[7] In a study of US recipients using Karnofsky score for adult recipients, over 80% of survivors reported the ability to function at 80% their functional status or greater at years 2 and 3.[7]
Although survival, functionality, and quality of life are enhanced post transplant, recipients are at increased risk of bronchiolitis obliterans syndrome (BOS) and other co-morbidities commonly seen with corticosteroids and chronic immunosuppression. The most common conditions include hypertension, chronic kidney disease, hyperlipidemia, and diabetes. According to ISHLT data, among 1-year survivors, 52.5% have hypertension, 26.3% have diabetes, 24.4% have kidney disease (1.7% on dialysis or requiring renal transplant), 24.2% have hyperlipidemia, and 9.6% have BOS. Among 5-year survivors, 84.4% have hypertension, 24.7% have kidney disease (3.3% on dialysis or requiring renal transplant), 56.5% have hyperlipidemia, 38% have diabetes, and 36.9% have BOS.[7] It is important for patients to realize that immunosuppression-related medical comorbidities of lung transplant are common, and that lung transplantation, therefore, provides a treatment for their lung disease that requires life-long management. Patients are often told that lung transplant provides a treatment but not a cure, meaning that patients will exchange their chronic lung disease for another medical condition that requires active therapy for the rest of their lives. Nonetheless, when a patient is willing to accept this possibility, and provided there are no major complications, lung transplantation provides a means to significantly improve one's quality of life.
CONCLUSIONS
Despite significant medical advancements, PAH remains a chronic, terminal disease. For patients who are refractory to medical therapy, lung transplantation remains the only therapeutic option. It is important to refer patients early to a transplant center, as the center can follow patients through their pre-transplant course of disease and better decide when to list for transplantation. Communication between the referring physician and transplant team is key in co-managing patients prior to and after transplantation. Given the technical challenges, it may be beneficial for lung transplantation for PAH to be performed in an experienced center. In the majority of cases, lung transplantation provides patients the chance to improve their quality and duration of life.