
Abstract
Pulmonary hypertension is characterized by cellular and structural changes in the walls of pulmonary arteries. Intimal thickening and fibrosis, medial hypertrophy and fibroproliferative changes in the adventitia are commonly observed, as is the extension of smooth muscle into the previously non-muscularized vessels. A majority of these changes are associated with the enhanced presence of a-SM-actin+ cells and inflammatory cells. Atypical abundances of functionally distinct endothelial cells, particularly in the intima (plexiform lesions), and also in the perivascular regions, are also described. At present, neither the origin(s) of these cells nor the molecular mechanisms responsible for their accumulation, in any of the three compartments of the vessel wall, have been fully elucidated. The possibility that they arise from either resident vascular progenitors or bone marrow-derived progenitor cells is now well established. Resident vascular progenitor cells have been demonstrated to exist within the vessel wall, and in response to certain stimuli, to expand and express myofibroblastic, endothelial or even hematopoietic markers. Bone marrow–derived or circulating progenitor cells have also been shown to be recruited to sites of vascular injury and to assume both endothelial and SM-like phenotypes. Here, we review the data supporting the contributory role of vascular progenitors (including endothelial progenitor cells, smooth muscle progenitor cells, pericytes, and fibrocytes) in vascular remodeling. A more complete understanding of the processes by which progenitor cells modulate pulmonary vascular remodeling will undoubtedly herald a renaissance of therapies extending beyond the control of vascular tonicity and reduction of pulmonary artery pressure.
INTRODUCTION
Pulmonary hypertension is characterized by cellular and structural changes in the walls of the pulmonary arteries. Intimal thickening and fibrosis, medial hypertrophy, and fibroproliferative changes in the adventitia are commonly observed, as is the extension of smooth muscle into previously non-muscularized vessels. A majority of these changes are associated with the enhanced presence of a-SM-actin+ cells and inflammatory cells.[1–4] Atypical abundances of functionally distinct endothelial cells, particularly in the intima (plexiform lesions) and also in the perivascular regions, are now described.[5,6] At present, neither the origin(s) of these cells nor the molecular mechanisms responsible for their accumulation in any of the three compartments of the vessel wall, have been fully elucidated.
Regarding their origin, it has been postulated that α-SM-actin-expressing cells accruing in systemic and pulmonary vascular lesions are exclusively derived from resident vascular smooth muscle cells (SMC) and / or from adventitial fibroblasts via ‘de-differentiation’ of the former or ‘differentiation’ of the latter [Figure 1]. Over the past decade, however, this concept has been expanded by new experimental data, suggesting many alternative sources of α-SM-actin-expressing cells (SM-like cells or myofibroblasts), in various vascular diseases, including pulmonary hypertension. For instance, the possibility that both epithelial and endothelial cells have the capability of transitioning into a mesenchymal, SM-like phenotype has been raised [Figure 1].[7–9] Resident vascular progenitor cells have been demonstrated to exist within the vessel wall, and in response to certain stimuli, to expand and express myofibroblastic, endothelial or even hematopoietic markers.[10–14] Multipotent resident lung vascular progenitors have also been demonstrated to reside within the Hoechstlow lung side population of the cells.[15,16] Bone marrow–derived or circulating progenitor cells have also been seen to be recruited to sites of vascular injury and to assume both endothelial and SM-like phenotypes [17–19] [Figure 1]. Here, we review the data supporting a contributory role for vascular progenitors (including endothelial progenitor cells, smooth muscle progenitor cells, pericytes, and fibrocytes) in systemic and pulmonary vascular remodeling. A more complete understanding of the processes by which progenitor cells modulate pulmonary vascular remodeling, will undoubtedly herald a renaissance of therapies extending beyond the control of vascular tonicity and reduction of pulmonary artery pressure.
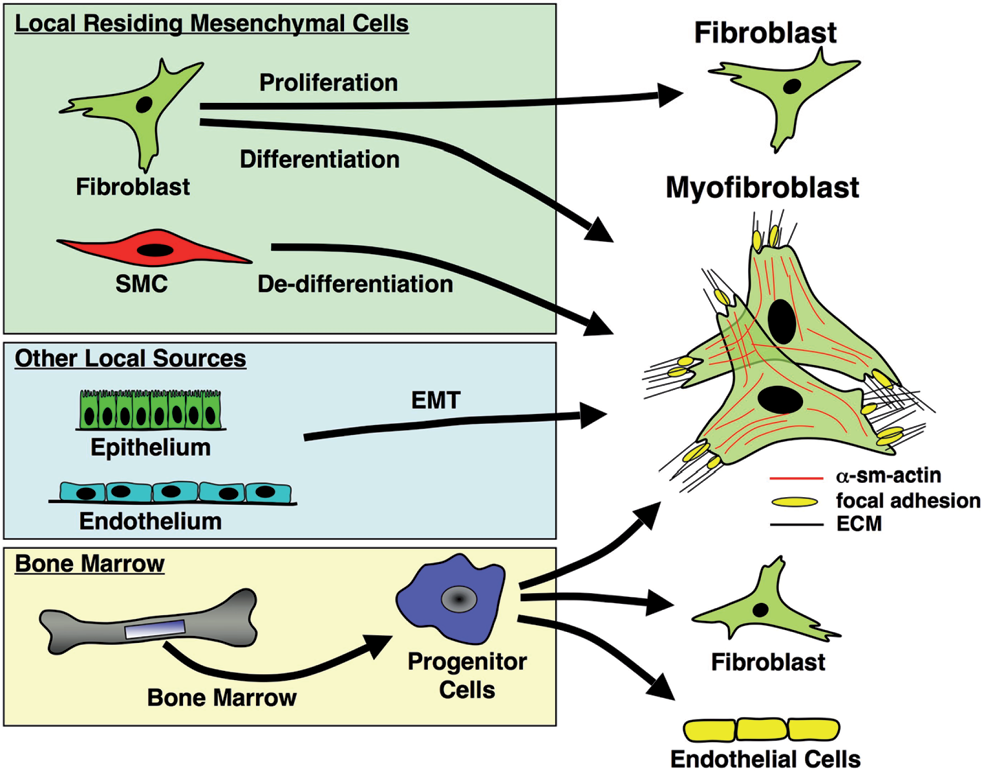
Potential origins of fibroblasts / myofibroblasts in the vessel wall. Several potential origins for tissue myofibroblasts have been proposed: (1) In several organs, α-SMA-expressing myofibroblasts are believed to originate from tissue-resident fibroblasts; (2) In the vasculature, myofibroblasts may arise through de-differentiation of resident SMC; (3) Epithelial cells can give rise to fibroblasts / myofibroblasts in the lung and other organs through a process of endothelial-mesenchymal transdifferentiation (EMT); (4) In the lung, endothelial-to-mesenchymal (EnMT) transition may provide another mechanism to generate myofibroblasts; (5) In various fibrotic lesions in tissue injury/repair processes, bone marrow-derived circulating progenitor cells are proposed, to contribute to the pool of myofibroblasts (Adapted from Hinz et al.) [8]
Origin and phenotypic characterization of circulating endothelial, smooth muscle, and fibroblast / myofibroblast (i.e., fibrocytes) progenitors
Circulating endothelial precursors
Currently, endothelial progenitor cells (EPCs) represent the most widely studied adult human progenitor cell subpopulation. Their discovery in 1997, by Asahara et al., marks a milestone in our understanding of the development of blood vessels in an adult.[20] EPCs are most commonly thought to be of bone marrow origin, but have recently been reported to also reside in the vascular adventitia,[21,22] and perhaps may also even exist in endothelium lining of blood vessels.[14,23–26] At present, however, the definition of EPC remains controversial and is not yet consistent.[27] Most commonly, marker combinations for identifying the putative circulating EPC comprise certain hematopoietic lineage markers, such as, CD133, CD34, VEGFR-2, Tie-2, and UEA-1 lectin.[24,25,27–29] However, recent studies have provided growing evidence supporting the inclusion of some circulating myeloid cells as functional EPCs, which can contribute to endothelial regeneration and ischemic or tumor angiogenesis. These cells are characterized by the expression of CD14; the subpopulations include CD14+ / CD34low, CD14+ / VEGFR-2+, CD14+ / VEGFR2+ / CXCR2+, and CD14low / CD16+ / Tie-2+.[30,31] Thus, it seems that circulating pools of functional EPCs correspond to a rather heterogeneous cell population of multiple origins, phenotypes, and tissue distribution. The common features of these cells, arguably, encompass the expression of the progenitor and endothelial markers (i.e., CD34, VEGFR2, CD31, VE-cadherin, von Willebrand Factor), colony forming capacity, and differentiation toward the endothelial lineage, with formation of angiogenic structures in vitro as well as in vivo.[25,27,30] The function of these EPCs in vivo is not known and may depend on the interplay with the inflammation and the tissue microenvironment. Investigators have also explored the growth properties and colony forming unit (CFU) potential of these cells, which have been found to be useful as biomarkers for outcomes in acute lung injury, as well as in cardiovascular diseases, including pulmonary hypertension[32,35]
Circulating smooth muscle precursors
Another vascular progenitor subtype, the smooth muscle progenitor cell (SPCs), has not been studied as intensively as the EPC. It has been shown that, similar to the EPCs, these progenitor cells can reside in the bone marrow, can circulate, or can be found in the peripheral tissues. Circulating SPCs can be distinguished by the expression of markers of mesenchymal / smooth muscle lineage, such as, endoglin (CD105), α-SM-actin (α -SMA), calponin, SM-myosin heavy chain (SM-MHC), SM22, or platelet-derived growth factor receptor-β (PDGFR-β).[36–39] Bone marrow-derived cells expressing smooth muscle markers have been observed in the remodeled intima of patients who have undergone sex-mismatched bone marrow transplants.[18,40] Furthermore, in a murine model, a subpopulation of sorted c-Kitneg / Sca-1+ / Linneg / PDGFR-β+ cells was reported to acquire the phenotype of mature SMCs in the presence of a platelet-derived growth factor-BB (PDGF-BB).[38] They are speculated to promote atherosclerotic plaque formation by producing extracellular matrix proteins.[39,41] Interestingly, similar to EPCs, human myeloid CD14+ SPCs have also been recently identified as a circulating CD14+ / CD105+ subpopulation.[37]
Circulating fibroblast precursors: fibrocytes
Another cell type that has received much attention as a potential vascular progenitor is the fibrocyte. Fibrocytes are bone marrow-derived mesenchymal progenitors that co-express hematopoietic stem cell antigens, markers of the monocyte lineage, and fibroblast products.[42–46] These cells produce extracellular matrix (ECM) components as well as ECM-modifying enzymes and can further differentiate into myofibroblasts both in vitro and in vivo, under permissive microenvironmental conditions.[43] Fibrocytes express the common leukocyte antigen CD45 as well as monocyte markers CD11a and b, and are variably reported to express CD14 and CD34. Upon stimulation, these cells express type 1 collagen, fibronectin, vimentin, and MMP-9. This expression pattern has led several investigators to hypothesize that fibrocytes, like some dendritic cell subsets, derive from precursors of the monocyte lineage. In fact, these cells express the major histocompatability complex class I and class II and the co-stimulatory molecules CD80 and CD86. Fibrocytes also exhibit antigen-presenting activity, and activate both the CD4 and CD8 T-lymphocytes.[43,46,47] The combination of collagen production and expression of CD45 (or one of the hematopoietic or myeloid antigens, CD11b, CD13 or CD34) is considered as sufficient criteria to discriminate fibrocytes from leukocytes, dendritic cells, endothelial cells, and tissue resident fibroblasts in vivo and in vitro. Fibrocytes can be distinguished from circulating or tissue resident mesenchymal stem / stromal cells, because the latter cells do express fibroblast products, but do not express CD34 or CD45, or the monocyte markers.[48]
Several factors effect the differentiation of fibrocytes from CD14+ monocyte precursors into mature mesenchymal cells. PDGF, interleukin (IL)-4, and IL-13 promote the differentiation of CD14+ mononuclear cells into fibrocytes.[49] By contrast, serum amyloid-P (SAP) inhibits the differentiation of CD14+ mononuclear cells into fibrocytes.[49,50] Recent studies also suggest that TH-1 derived cytokines such as IL-12 and interferon-γ may inhibit fibrocyte differentiation. On the other hand, TH-2 products, such as IL-4 and IL-13, stimulate fibrocyte differentiation as does TGF-β. Further differentiation into cells, ultrastructurally, phenotypically, and functionally similar to mature fibroblasts and / or myofibroblasts is promoted by stimulation with TGF-β or endothelin.[49] The resulting cell population produces more collagen and fibronectin than the relatively immature fibrocyte. In certain circumstances, the fibrocyte begins to express α-SM-actin, while downregulating the leukocyte marker expression, and assumes a myofibroblast phenotype.
Resident endothelial, smooth muscle, and fibroblast / myofibroblast progenitors
Resident intimal progenitors
In addition to a circulating source of EPCs and mesenchymal cells, there is increasing evidence to suggest that cells with progenitor characteristics exist within the vessel wall. Pioneering research uncovering endothelial cell division and proliferation by tritiated thymidine uptake is now nearing its fortieth anniversary.[51] A few years later, it was determined that endothelial proliferation could increase during injury and that the replicating endothelial cells appeared in clusters termed ‘high turnover regions’.[52] Following the discovery of circulating EPCs, it was demonstrated that EPCs exist in the wall of the human embryonic aorta, which can differentiate into mature endothelial cells and form vascular-like structures under in vitro conditions.[26,53] Further studies revealed the presence of a complete hierarchy of EPCs in the wall of the human adult blood vessels as well as umbilical cord.[26,54] Recently, it was shown that large- and middle-sized human arteries and veins in several organs contain EPCs in a distinct zone of the vascular wall (termed ‘vascular wall-resident EPCs’, VW-EPCs), and that the region was named the ‘vasculogenic zone’ [Figure 2]. This zone was located between the outer media and the adventitial layers.[10,14] Interestingly, the VW-EPCs were reported to differentiate not only into mature endothelial cells, but also into hematopoietic and local immune cells such as macrophages. In the vessel wall these cells were not CD34+, but CD31−, and they also expressed VEGFR-2 and Tie-2. Only a few cells in this zone of the vascular wall were positive for a leukocytic antigen CD45. Most intriguingly, recent findings suggested that some circulating EPCs might in fact derive from the intimal vascular endothelial layer,[54] perhaps in regions such as the ‘vasculogenic zones’. If confirmed, such a phenomenon would make unclear the current distinctions between resident and circulating progenitors, and potentially also between the processes of angiogenesis and vasculogenesis.
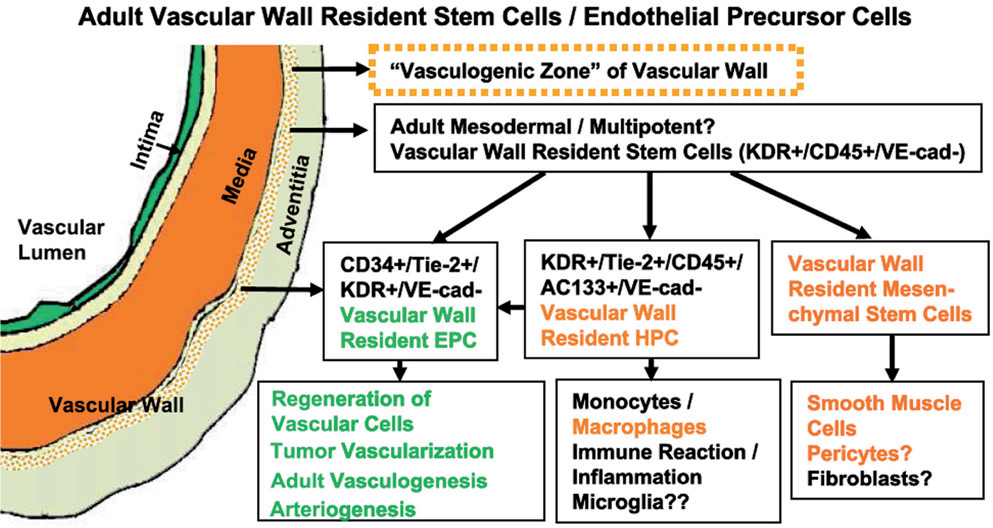
Hypothetical scheme illustrating the concept of the ‘vasculogenic zone’. This vascular mural zone at the border between the media and adventitia contains EPCs and probably also multipotent mesodermal stem cells. EPCs present in this zone are proposed to differentiate into endothelial cells and form capillary-like sprouts from the vascular wall, whereas, the multipotent mesodermal stem cells in this zone may serve as precursors of macrophages, fibroblasts, and SMC (Adapted from Zengin et al.)[14]
Resident adventitial progenitors
Other investigators have reported that non-EPC progenitor cells reside in the adventitia of the vessel wall and are capable of giving rise to vascular wall cells including SMCs. Hu et al. reported that stem cell antigen-1 (Sca-1)+ cells, with a potential to differentiate into SMC, reside in the aortic adventitia of adult mice.[55] Subsequent studies by Passman et al. have confirmed the presence of Sca-1+ progenitor cells at the medial-adventitial border. In vivo, these adventitial Sca-1+ cells do not express SMC marker proteins. They do, however, express transcription factors thought to be required for SMC differentiation, including the serum response factor (SRF) and myocardin family members, and in vitro they readily differentiate into SM-like cells. It is possible that these cells, when activated in response to injury, contribute to the accumulation of SM-like cells in the vessel wall.[56]
Resident medial / adventitial border progenitors
In addition to progenitors located within distinct medial or adventitial compartments, studies have suggested that there exists a progenitor cell subset at the medial / adventitial border, in the medium to large arteries and veins. This CD34+ / CD31-cell type can apparently give rise to KDR+ / Tie2+ endothelial cells and capillary sprouts.[10,14] Interestingly, additional cells within these ‘vasculogenic zone’ cells are CD45+, pointing to the possibility of a vascular wall resident HSC population, at least in the human internal thoracic artery segments studied. Strengthening these findings, Pasquinelli et al. have determined that the thoracic aorta segments contain a mesenchymal stromal cell (MSC) population capable of differentiation into the endothelium.[57] Of late, this group has provided evidence that MSCs within the vasculogenic zone are morphologically and immunophenotypically stem-like. By flow cytometry, MSCs can be identified by the expression of mesenchymal antigens CD29, CD44, CD90, CD73, CD105, and CD166, and lack of expression of the hematopoietic lineage markers CD45 and CD34.[58] In addition, the vasculogenic zone MSCs robustly express ‘stemness’ markers OCT-4, Notch-1, and Stro-1, and exclude the Hoechst dye, as mentioned a little further in the text, for other SP cells. Further confirmation using robust lineage-tracking systems is needed to fully elucidate the true potential of progenitor cell subsets within the vasculogenic zone. Nevertheless, it is conceivable that in medium-to-large-sized vessels, the wall itself contains the progenitor cell potential for renewal of all vessel cell types, and possibly immune and / or inflammatory cell types.
Pericytes as resident progenitor cells
Pericytes were discovered over one hundred years ago [59,60] and are subendothelial cells resident in large vessels down through to the microvasculature. Currently, the true nature of their origin remains complex and poorly understood. Their location in vessels can vary, from a peri-endothelial localization to the media, adventitia, and in association with the vasa vasorum.[61] They are best characterized in the microvasculature, where they play important roles in capillogenesis, microvascular tone, as well as providing structural integrity.[62] New investigations into these enigmatic cells have revealed a possible progenitor cell role. Although it has been known for some time that pericytes are able to differentiate into osteoprogenitor cells,[63] they seem to figure prominently in aortic calcification during atherosclerosis.[64] The characteristic expression pattern of pericytes has been reviewed elsewhere,[65] but a thorough characterization of the specific pericyte markers associated with the bona fide multipotent progenitor potential remains to be defined. Interestingly, it has been suggested that pericytes and vasculogenic zone MSCs may be closely related.[65] The distribution of pericytes throughout the vasculature suggests that perhaps pericytes or MSCs are vascular progenitor cells that are competent to renew a variety of cell types, while simultaneously maintaining a ‘lineage-allegiance’[65] responsive to the needs of the tissue of residence. A better understanding of the mechanisms of control that facilitate pericyte (and vessel wall MSC) progenitor competence, particularly in the context of vascular remodeling, could open a new era of therapy targeting the vessel wall to repair itself.
Lung side population cells as progenitors of endothelial and mesenchymal cells
Another source of resident lung vascular progenitors, which may play a role in the pathogenesis of pulmonary arterial hypertension, is the Hoechstlow CD45neg side population of cells, also termed as ‘SP’. The term side population (SP) is based on the cytometric profile in which there is a side arm of cells protruding from the main Hoechst stained population and is referred to as Hoechstlow [Figure 3]. The Hoechst vital dye is taken up by live cells and fluoresces in red and blue when excited by a UV laser. An ABCG2 multidrug resistance (MDR) transporter mechanism allows the cells to pump out the dye, thus leading to a lower fluorescence intensity.[66,67] ABCG2 as well as other MDR transporters are believed to be a hallmark of primitive cell types, HSC, and cancer cells.[66–71] The murine lung SP cell populations (Hoechstlow CD45neg) have been isolated and characterized in vitro as an enriched tissue-specific source of organ-specific pulmonary precursors, having mesenchymal stem cell properties as well as both endothelial and epithelial lineage potential.[15,16,72–75] The origin of lung SP was examined by bone marrow transplantation analyses and it was determined that the CD45pos fraction was derived from the bone marrow, while the origin of the CD45neg population was undefined.[75] These studies form the basis for the identification of the SP, or ABCG2, as a resident adult tissue-specific stem cell marker.
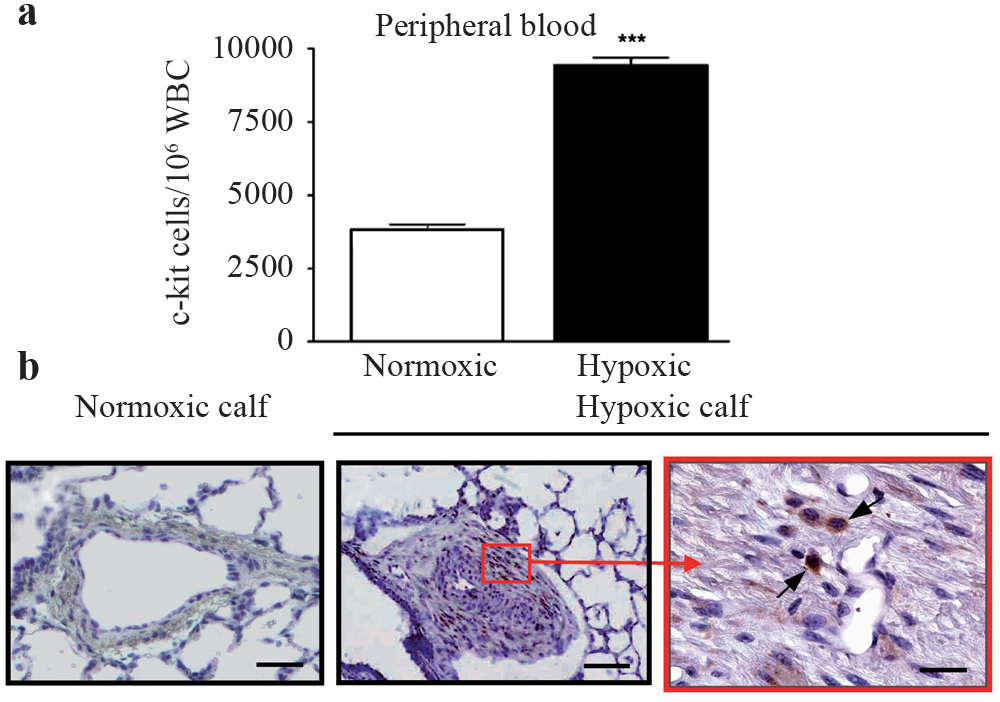
Identification of increased markers of c-kit+ cells in the blood and pulmonary artery of chronically hypoxic animals. Increased numbers of c-kit+ cells are present in the blood of chronically hypoxic calves compared to controls. Immunohistochemistry (brown peroxidase signal) revealed greater number of c-kit+ cells (arrows) in the vessel wall of the distal PA from hypoxic animals compared to controls. The c-kit+ cells were localized, contiguous to, and within, the vasa vasorum in the adventia (Adapted from Davie et al.) [106]
The vascular potential of the lung SP is defined in vitro by their ability to express VE-Cadherin, bind isolectin B4, take up diLDL, and their ability to form angiogenic tubes in matrigel.[15] More recent studies defined the mesenchymal stem cell potential of this Hoechstlow CD45neg in both mixed populations of cells as well as with single cell clones. These cells express high levels of telomerase, relative to the liver tissue, which does not decrease significantly over time.[16] These cells express characteristic mesenchymal markers (CD44, CD90, CD105, CD106, CD73, and Sca-I) and lack any hematopoietic markers (CD45, c-kit, CD11b, CD34, and CD14). The clonal analyses demonstrated that a single lung SP could assume the phenotype of mesenchymal lineages such as bone, cartilage, and fat.[16] Although the true role of these cells in vivo is yet to be determined, as this cell population has both vascular endothelial and mesenchymal stem cell characteristics, it is likely that they differentiate to endothelial, smooth muscle, and myofibroblast lineages, depending on the microenvironment. Thus, the lung SP may play a role in the remodeling associated with pulmonary hypertension.
Contribution of circulating vascular progenitors to systemic vascular remodeling
The contribution of circulating progenitor cells to vascular remodeling has been studied more extensively in systemic vascular disease than in pulmonary vascular disease. Many of these studies serve as the rationale for investigations in pulmonary circulation, and thus, a brief review of this study is critical for a better understanding of progenitor cells in pulmonary vascular remodeling.
As mentioned earlier, in contrast to the initial conventional assumption that damaged organs are repaired only by migration and proliferation of adjacent resident cells, accumulating the evidence supports the idea that multifunctional progenitor cells are mobilized into the circulation and are recruited specifically into the sites of tissue regeneration. Many reports have demonstrated that bone marrow-derived EPCs significantly contribute to neovascularization and re-endothelialization after acute vascular injury.[20,76] Furthermore, several animal and clinical studies have demonstrated that transplantation of autologous EPCs or unfractionated bone-marrow cells is effective for the treatment of ischemic cardiovascular disease.[19,77] On the other hand, there is increasingly good evidence demonstrating that bone marrow cells or circulating progenitor cells can participate not only in the maintenance or restoration of vascular homeostasis, but also in the pathogenesis of various vascular diseases. The role of EPC cells in vascular healing has been well demonstrated and recently reviewed and will not be discussed further.[78,79] However, the role of progenitor cells in pathological angiogenesis and intimal lesion formation is briefly reviewed here, because of its relevance to the mechanisms involved in vascular remodeling in pulmonary hypertension.
Angiogenesis has been implicated in the pathogenesis of a variety of systemic disorders, including diabetic retinopathies, tumors, rheumatoid arthritis, and cirrhosis.[80,81] EPCs have been shown to contribute to the pathological angiogenesis observed in these conditions.[11,82,83] Tumor angiogenesis, in particular, is associated with the recruitment of hematopoietic and circulating EPCs.[84] In fact, experimental evidence has demonstrated that hematopoietic progenitors expressing vascular endothelial growth factor receptor-1 (VEGFR1) are required for regulation of tumor metastasis.[85] In this manner, injection of bone marrow cells promoted injury-associated retinal angiogenesis.[86] Furthermore, endothelial cells that accumulate in the neointimal lesions in allografts and originate from circulating and bone marrow-derived progenitors, are responsible for the formation of microvessels in the setting of transplant atherosclerosis.[11] These observations raise the possibility that transplantation of endothelial progenitor or bone marrow cells may, under certain circumstances, promote tumor formation, diabetic retinopathy, and atherosclerosis, by augmenting disease-associated pathological angiogenesis. However, some clinical studies have demonstrated that the number of circulating EPCs correlates inversely with the risk factors of coronary artery disease.[33] In addition, the levels of circulating EPCs have been reported to predict the occurrence of cardiovascular events.[87] These observations have prompted some investigators to suggest that the physiological levels of circulating EPCs, in fact, function to prevent atherosclerosis, without promoting unfavorable angiogenesis.
The observations that a majority of cells in neointimal lesions express certain SM-markers (typically α-SM-actin [88]) has led to the general assumption that SMCs in the adjacent medial layer migrate into the subendothelial space, and proliferate and synthesize the extracellular matrix, thereby contributing to neointimal formation.[89] However, many recent pathological studies have demonstrated that neointimal SM-like cells are phenotypically distinct from medial SMCs. For example, unlike SMCs in the normal vascular media that express a differentiated contractile phenotype, neointimal SM-like cells are characterized by a large number of synthetic and secretory organelles. These ‘synthetic’ SM-like cells secrete extracellular matrix components and express lower levels of SM-specific contractile proteins.[90] These synthetic SM-like cells proliferate and migrate significantly in response to various growth factors present in the injured vessel wall.[91] In fact, much effort has been devoted to understanding the regulators of the vascular SM phenotype that control the transition from a quiescent differentiated SMC, under normal conditions, to proliferative de-differentiated SM-like, in the presence of pathological stimuli. Intriguingly, however, it was reported that some neointimal SM-like cells express a number of hematopoietic lineage makers along with certain SM markers.[92] Macrophage-like SM-like cells with phagocytic activity have been obtained from human arteries.[93] Although it has been assumed that resident medial SMC ‘transdifferentiate’ into macrophage-like cells, recent studies have demonstrated that some ‘synthetic’ SM-like cells with characteristics of macrophages, are derived from circulating blood cells rather than from medial SMC.[22,94] Taking advantage of both genetically modified mice as well as the use of a chimeric mouse model of parabiosis, recent studies have demonstrated convincingly that in a variety of systemic vascular injuries, bone marrow-derived cells home to the damaged vessels and contribute to both vascular repair and pathological remodeling, by differentiating into cells expressing mesenchymal or SMC characteristics.[22,95] Smooth muscle progenitor cells may in fact contribute to plaque formation through the production of extracellular matrix proteins.[39]
In addition to EPCs and SM progenitor cells, fibrocytes have also been described as being present in the diseased systemic vessel wall. Fibrocytes (procollagen-1+ / CD34+) have recently been identified in the fibrous cap of human atherosclerotic lesions.[96,97] Moreover, subendothelial aSMA+ myofibroblasts, co-expressing CD68 or CD34, have been found in the lipid-rich areas of the atherosclerotic intima in the human aorta and in the fibrous cap of human carotid arteries, suggesting that these cells are fibrocytes.[96,98] Furthermore, one of the subsets of monocytes that is preferentially recruited into the arterial wall during experimentally induced arteriosclerosis in mice, represents the murine counterpart of the human inflammatory subset of CD14+ / CD16neg mononuclear cells, which express the MCP-1 receptor, CCR2.[99] These observations are consistent with the elegant new studies from Varco et al. demonstrating that intimal hyperplasia in an ovine carotid artery patch graft model is partially due to hematopoetic circulating progenitor cells, fibrocytes that acquire mesenchymal features as they mature at the site of injury.[100] Thus, a CD14+ / CD16− / CCR2+ subpopulation may be involved in atherogenesis and may contain fibrocyte precursors that contribute to lesion formation. In addition to diseases of the proper vessels, fibrocytes are associated with pathological vascular remodeling in the context of a diverse set of disease states including obliterative bronchiolitis,[101] asthma,[102] pulmonary fibrosis,[103] and ischemic cardiomyopathy.[104] Collectively, these observations are supported by epidemiological data showing that MCP-1 plays a major role in the pathogenesis of atherosclerosis and disease progression, and may promote recruitment of fibrocytes to the vessel wall.[105]
Contribution of circulating vascular progenitors to pulmonary vascular remodeling
Animal studies
Based on the observations in systemic vascular diseases, Davie et al. were among the first to examine the possibility that circulating progenitor cells contribute to pulmonary vascular remodeling.[106] They tested the hypothesis that hypoxia would stimulate mobilization of bone marrow (BM)-derived c-Kit+ cells (c-Kit is a transmembrane tyrosine kinase receptor for the stem cell factor and a generally accepted marker for BM-derived hematopoietic stem cells (HSC)) into the circulation, and create an environment, specifically in the pulmonary artery, which facilitates circulating c-Kit+ cell adhesion, and thus, increase the number of c-Kit+ cells within the remodeled pulmonary artery. The study demonstrated that chronic hypoxia significantly increased the number of c-Kit+ cells in the circulation and decreased their numbers in the BM, thus supporting the previously reported idea that ischemic stimuli led to the mobilization of BM-derived stem cells [Figure 3A].[106–107] Of relevance is the fact that genes regulated by hypoxia, including erythropoietin and VEGF, have been implicated in the generation and differentiation of hemangioblasts, the precursors of both HSC and primitive endothelial cells.[108] Moreover, VEGF has been shown to mediate the mobilization of BM-derived HSCs and EPCs, which promote tumor blood vessel growth.[84] An increase in the number of c-Kit+ cells in the remodeled adventitia as well as in the expanding vasa vasorum of hypoxic pulmonary arteries compared with the relatively acellular pulmonary adventitia of normoxic animals was also observed [Figure 3]. Elevated expression of proteins that serve critical roles in recruitment, retention and differentiation of progenitor cells (such as VEGF, fibronectin, thrombin, and osteopontin) were observed in regions enriched in c-Kit+ cells. Each of these molecules has been implicated in inflammatory cell responses, including adhesion, migration, division, and differentiation of cells, including monocytes, macrophages, and progenitor cells. Indirect evidence that the c-Kit+ cells might contribute to the expansion of the vasa vasorum and accumulation of fibroblasts / myofibroblasts in the vessel wall was provided by in vitro observations that peripheral blood MNCs derived from hypoxic calves exhibited the potential to differentiate into endothelial as well as SM-like cell phenotypes, depending on the culture conditions.
In this study, it was also demonstrated that a marked expansion of the vasa vasorum occurred in the setting of chronic hypoxic pulmonary hypertension [Figure 4], as based on cell localization studies [Figure 4C] and studies in systemic vascular disease, the expanding vasa vasorum acted as a conduit for delivery of inflammatory leukocytes and progenitor cells (c-Kit+) to the vessel wall.[98] Shortly after publication of these data came other reports supporting the idea that circulating progenitor cells contribute to remodeling in various models of pulmonary hypertension. Hayashida et al. used green fluorescent protein-BM-transplanted (GFP-BMT) chimeric mice to investigate the possible role of BM-derived cells in hypoxia-induced pulmonary vascular remodeling.[109] These investigators demonstrated that hypoxia induced the recruitment of significant numbers of GFP+ BM-derived cells into the pulmonary artery wall, including the adventitia. Very few cells were observed in the control mice. The investigators quantified the number of GFP+ cells that co-expressed α-SMA in the lung tissues of both the control and pulmonary hypertensive mice and found significant increases in the numbers of GFP+ / α-SMA+ cells in the pulmonary media and adventitia. They found that the number of GFP+ / α-SMA+ cells increased with time in parallel with the progression of pulmonary hypertension. Collectively, the Davie et al. and Hayashida et al. studies provide evidence that BM-derived cells, which are mobilized and accumulate in the pulmonary arteries, contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension.[106,109]
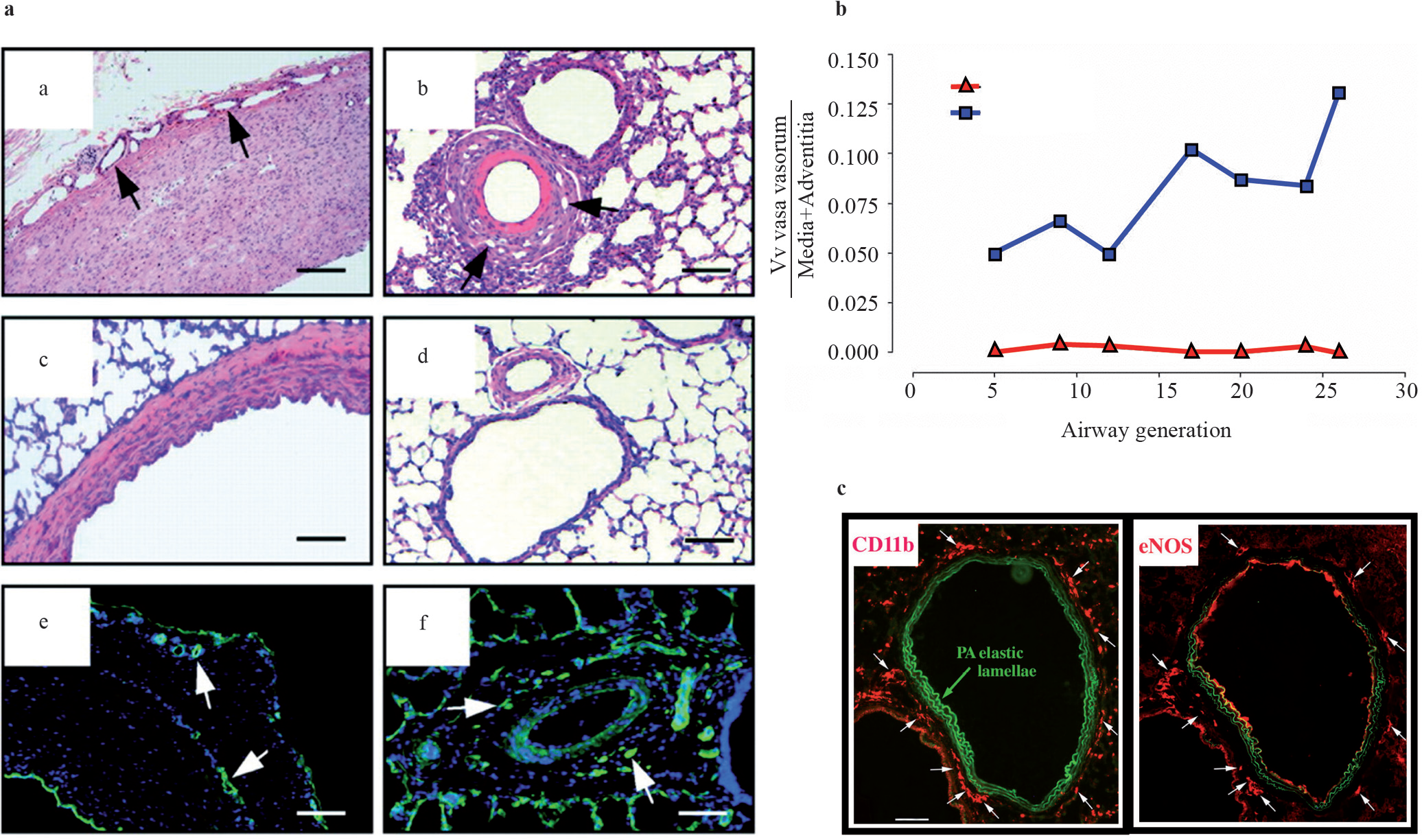
Expansion of the vasa vasorum in hypoxic pulmonary hypertension. Increase in the density of vasa vasorum (arrows) in proximal (a) and distal (b) vessels from hypoxic animals, compared with proximal (c) and distal (d) vessels from normoxic animals. Vasa (arrows) in hypoxic proximal and distal arteries express platelet endothelial cell adhesion molecule (PECAM)-1 (e and f, respectively). Quantitative morphometric analysis demonstrated that the volume density (Vv) of the vasa vasorum is significantly greater in the adventitia and media of the pulmonary arteries of chronically hypoxic animals, at every level along the longitudinal axis of the pulmonary circulation than in the controls. Monocytes (CD11b) appear preferentially next to the vasa (eNOS+cells) in hypoxic vessels
Subsequently, additional reports have emerged supporting the role of specific progenitor cells, including fibrocytes and bone marrow-derived mesenchymal progenitors, in the pulmonary vascular remodeling process. Circulating fibrocytes have been implicated in the pathogenesis of lung fibrosis in several mouse models, including irradiation- and bleomycin-induced lung injury.[102,103,110,111] Fibrocytes have also been shown to contribute to the subepithelial fibrosis observed in the airways of mice with experimentally induced asthma.[112] In experimental animal models (rat and neonatal calf) with pulmonary hypertension, Frid et al. reported that exposure of these animals to chronic hypoxia led to a robust accumulation of fibrocytes in the adventitia of pulmonary, but not systemic vessels.[17] Using confocal microscopy, the investigators found that at least 40% of the monocyte-like cells accumulating in the pulmonary artery adventitia co-expressed the leukocytic marker CD45 and the mesenchymal marker, type 1 pro-collagen, consistent with a fibrocyte phenotype [Figure 5]. A significant but lesser proportion of cells co-expressed CD45 and α-SM-actin, thus indicating the transition of the recruited circulating fibrocytes toward the myofibroblast phenotype [Figure 6]. In vivo labeling of circulating monocytic cells with liposome-encapsulated DiI (a red fluorochrome) provided additional evidence that, in response to chronic hypoxic exposure, the circulating cells were recruited specifically to the pulmonary vessels and not to the systemic vessels, and consequently expressed fibroblast antigens (type 1 procollagen) in the remodeled pulmonary vessel wall. Most importantly, the investigators demonstrated a causal link between fibrocyte accumulation and vascular wall remodeling, by showing that depletion of circulating monocytes / fibrocytes, using two different strategies (gadolinium chloride- and chlodronate-encapsulated liposomes), abrogated hypoxia-induced perivascular remodeling in the rat model of hypoxic pulmonary hypertension. These results are consistent with studies in other organ systems, where inhibition of fibrocyte accumulation resulted in reduced collagen deposition and reduced accumulation of myofibroblasts.[100,104,113,114] However, it must be noted that fibrocytes themselves are pro-inflammatory cells and produce a number of cytokines and growth factors that can induce angiogenesis, fibroblast hyperplasia, and release of ECM molecules from the resident tissue fibroblasts.[115] Therefore, the correlations between fibrocyte accumulation and tissue remodeling may also reflect the paracrine effects of these cells on resident vascular wall cells.
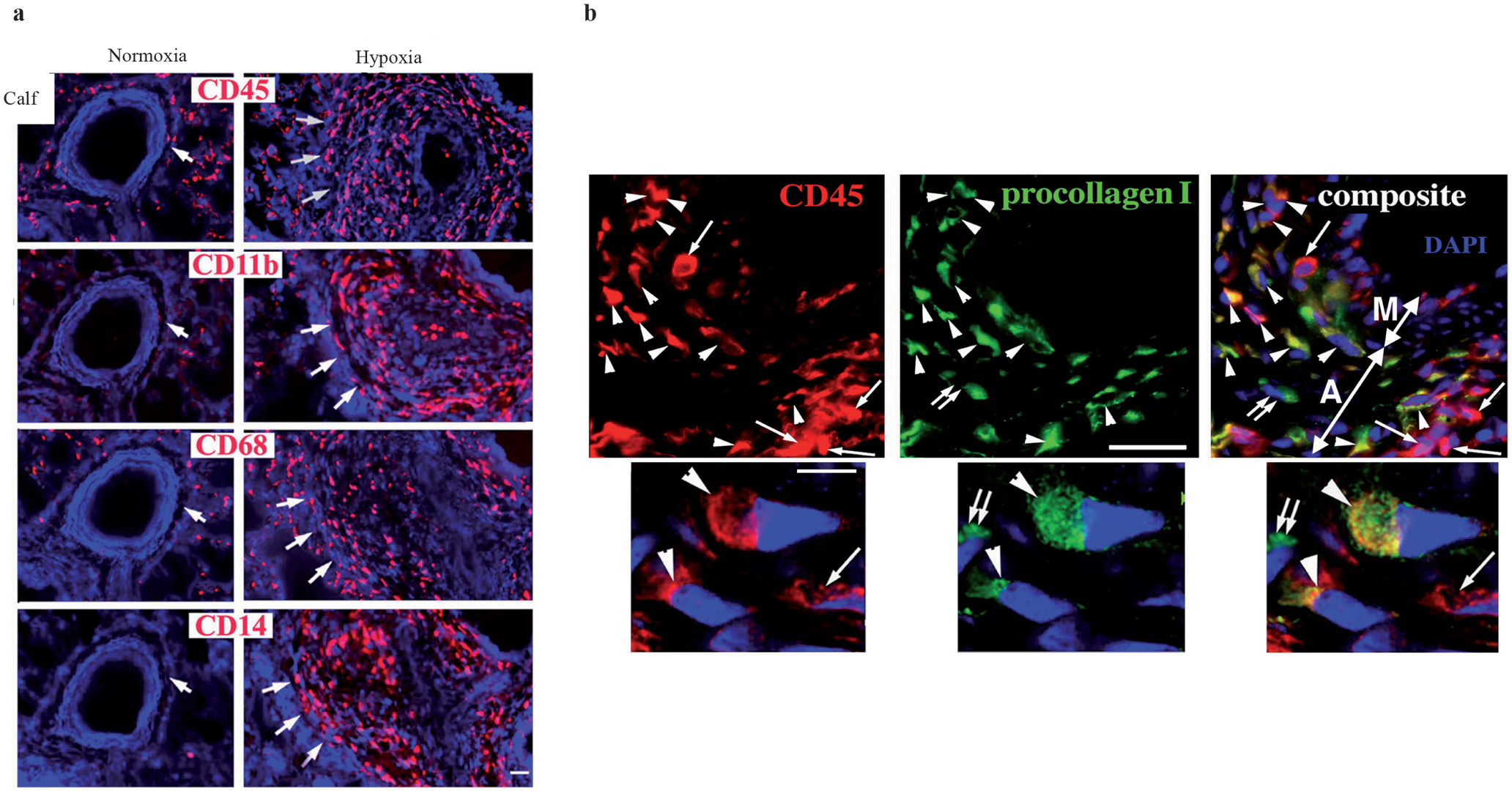
Hypoxia-induced pulmonary remodeling is characterized by monocyte/fibrocyte accumulation in the adventitia and media. (a) Sustained hypoxia induces a robust appearance of mononuclear CD45+ cells (indirectly labeled with red fluorochrome) in the pulmonary arterial adventitia (these lesions are marked by triple large arrowheads in the ‘Hypoxia’ columns). (b) A large proportion (around 40%) of the CD45 + leukocytic cells (red fluorescence) accumulating in the remodeled pulmonary arterial adventitia of hypoxic hypertensive calves co-express type 1 pro-collagen (green fluorescence), suggestive of a fibrocyte phenotype. Scale bars, 5 μm. (Adapted from: Frid et al.).[17]
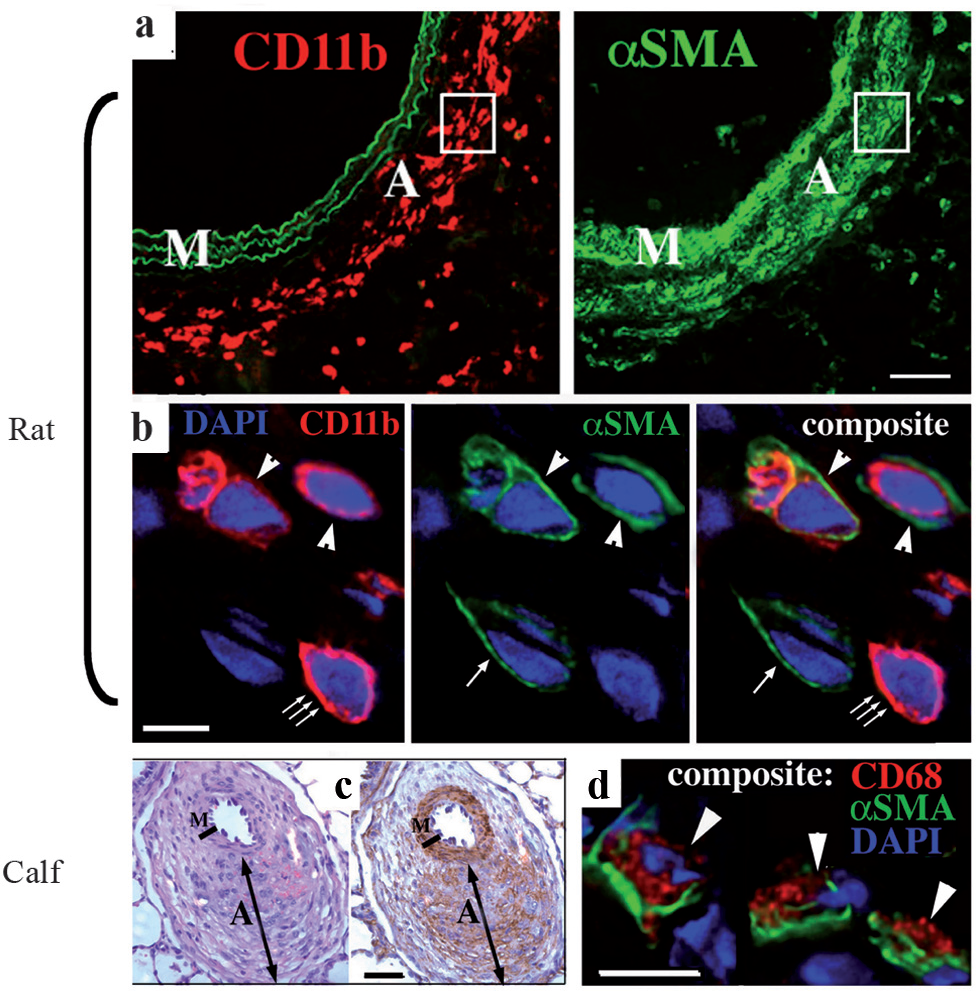
Hypoxia promotes the emergence of cells co-expressing leukocytic (CD11b) and the myofibroblast marker α-SM-actin. (a) A robust influx of CD11b+ cells (red) in the PA adventitia of chronically hypoxic rats correlates with a significant number of α-SMA+ myofibroblasts in that region. A – Adventitia; M – Media. (b) Double-label immunofluorescent staining for CD11b (red) and α-SMA (green), followed by deconvolution confocal microscopy analysis of the ‘boxed’ area in A, demonstrate cells that co-express both markers (arrowheads), or express only one of the markers (α-SMA, single arrow or CD11b, triple arrows). (c) Significant numbers of α-SMA+ myofibroblasts (brown staining in the adventitia (labeled (‘A’)) are present in distal PAs of the chronically hypoxic calves. (d) Double-label immunofluorescent staining, followed by deconvolution confocal microscopy, demonstrate that some of the α-SMA+ myofibroblasts (green) co-express a macrophage marker CD68 (red) (arrowheads). DAPI (blue) labels cell nuclei. M, media; A, adventitia. Scale bars: 50 μm (a, c); 5 μm (b, d). (Adapted from: Frid et al.)[17] cells from these tissues were able to give rise to adipocytes, and underwent osteogenic differentiation. The progenitor cells typically co-expressed the intermediate filaments vimentin and smooth muscle alpha-actin, suggesting that they had identified a myofibroblast cell predominant in the endarterectomized tissue.
A recent study on two distinct rodent models of pulmonary hypertension highlights the potential contribution of BM-derived progenitor cells and the efforts to unravel their contribution to pulmonary vascular remodeling. Angelini et al., using GFP + transgenic mice, determined that chronic hypoxia caused the recruitment of cells positive for Sca-1 and c-kit, but negative for CD31 and CD34, to neovascularized small vessels.[116] Recent studies by Spees et al. examined the potential, specifically of the bone marrow of BM-derived progenitor cells, to contribute to the repair and remodeling observed in the lung and heart during monocrotaline (MCT)-induced progressive pulmonary hypertension.[117] Female rats transplanted with GFP+ male (Y-chromosome+) BM cells were used as models. In the control, in the untreated GFP+-BMT chimeric rats, approximately 15% of the cells in the lung were GFP+, whereas, in the MCT-treated chimeric rats, the number of GFP+ cells increased to 35-45%. The pulmonary GFP+ cells in both cases were a mix of hematopoietic inflammatory and non-hematopoietic cells. A large number of BM-derived GFP+ cells in the lung were fibroblasts and myofibroblasts and thus may have contributed to lung fibrosis following irradiation and MCT treatment. The possibility that cell fusion could have been playing a role in the differentiation of BM-derived cells into mesenchymal cells was addressed. However, as indicted by fluorescence in situ hybridization (FISH) assays, the incidence of cell fusion in the lung was very low, ruling this out as a major contributing mechanism. Importantly, the investigators also showed the presence of BM-derived cells in the right ventricle of MCT-treated chimeric rats. Therefore, as in the case of many fibrotic diseases, these results identify BM-derived progenitors as important players in vascular pathology and suggest that strategies aimed at the inhibition of the recruitment of these cells may constitute important therapeutic options in the treatment of pulmonary vascular disease.
Human studies
Newly emerging data in humans with PH support the role of progenitor cells in the vascular remodeling that characterizes these patients. Patients with chronic obstructive pulmonary disease (COPD) often exhibit striking vascular remodeling in the pulmonary muscular arteries in precapillary vessels. The hallmark of remodeling in this group of patients is the thickening of the intima due to the accumulation of SM-like cells. Peinado et al. presented evidence that CD133+ EPCs participate in this process.[118] They demonstrated an increased number of CD133+ cells infiltrating the hyperplastic intima of pulmonary arteries very close to the areas of the denuded endothelium. The number of recruited progenitor cells correlated with the thickness of the arterial wall, suggesting a potential association with the severity of the remodeling process.[118] Subsequently, in vitro studies by the same group demonstrated that the CD133+ cells might acquire the morphology and phenotype of not only the endothelial cells, but also the SM-like cells. CD133+ cells, co-incubated with the isolated human pulmonary artery, were shown to migrate into the intima and differentiate into SMC. Progenitor cell differentiation was produced without fusion, with mature SMC. Thus, CD133+ cells exhibit a certain degree of plasticity, with an ability to differentiate into both endothelial and SM-like cells, which re-enforces the idea regarding their potential role in pulmonary hypertension in COPD.
Firth et al., reported that endarterectomized tissues from patients with chronic thromboembolic pulmonary hypertension (CTEPH) contained mesenchymal progenitor cells that were CD44 (+) CD73 (+), CD90 (+), CD166 (+); > 80% CD29 (+); 45-99% CD105 (+); CD34 (-), and CD45 (-).[119] Furthermore, they showed that sorted progenitor cells from these tissues were able to give rise to adipocytes, and underwent osteogenic differentiation. The progenitor cells typically co-expressed the intermediate filaments vimentin and smooth muscle alpha-actin, suggesting that they had identified a myofibroblast cell predominant in the endarterectomized tissue.
Evidence of endothelial progenitor dysfunction has recently been provided by Toshner et al. In this study, the endothelial cells within the plexiform lesions were determined to express CD31, CD133, c-kit, CXCR4, and SDF-1.[120] Furthermore, fluorescence-activated cell sorter (FACS) analysis showed an increase in CD133 + CD34 + VEGFR2+ circulating angiogenic progenitors in PAH compared to controls. In addition, late-outgrowth endothelial progenitor cells from patients with PAH demonstrated increased proliferation, migration, and angiogenic capacity compared to controls. This study points to the significant contribution of circulating progenitors to vascular remodeling, most particularly development of endothelial cells containing obliterative lesions.
It has also been recently reported that the numbers of apoptosis-resistant, highly proliferative circulating CD133+ cells increase in human idiopathic PAH.[5,121] The investigators raised the possibility that these cells contributed to the intimal lesions observed in these patients. Importantly, a recent study by Majka et al. demonstrated increased numbers of CD133+ cells in and around the pulmonary vascular lesions in both idiopathic PAH (iPAH) and familial PAH (fPAH)2 [Figure 7]. The increases in CD133+ and CD45+ cells raised questions as to how these cells affected resident cell function and the remodeling process in general. One possibility was that cell–cell fusion events were present and contributed to the remodeling process. Cell fusion could result in genetic changes, which conferred selective growth advantages as well as microsatellite instability, point mutations, allelic imbalance, and loss of heterozygosity.[122–124] The result of such changes were uncoupling from normal checkpoint apoptosis, germline mutation, and hypermethylation, which could, in part, explain the presence of the apoptosis-resistant cells in PAH. Majka et al. therefore analyzed ploidy in cells comprising vascular lesions, as any cell fusion event would result in at least twice the chromosomal content (4N)2. The investigators did not detect any chromosomal content amplification in either iPAH or fPAH specimens, thus suggesting that genetic instability and proliferative advantage of cells in the hypertensive tissues were not the result of aneuploidy. Thus, it was speculated by the authors that if a neoplastic ‘switch’ had occurred in the cells that characterized the vascular lesions in PAH, it did not involve genes typical to those usually observed in lung cancer.[125,126] The possibility that fibrocytes were also present in the remodeled vessel wall of patients with severe PAH was raised, although not demonstrated as conclusively as was their presence and contribution to airway remodeling in patients with asthma.[112]
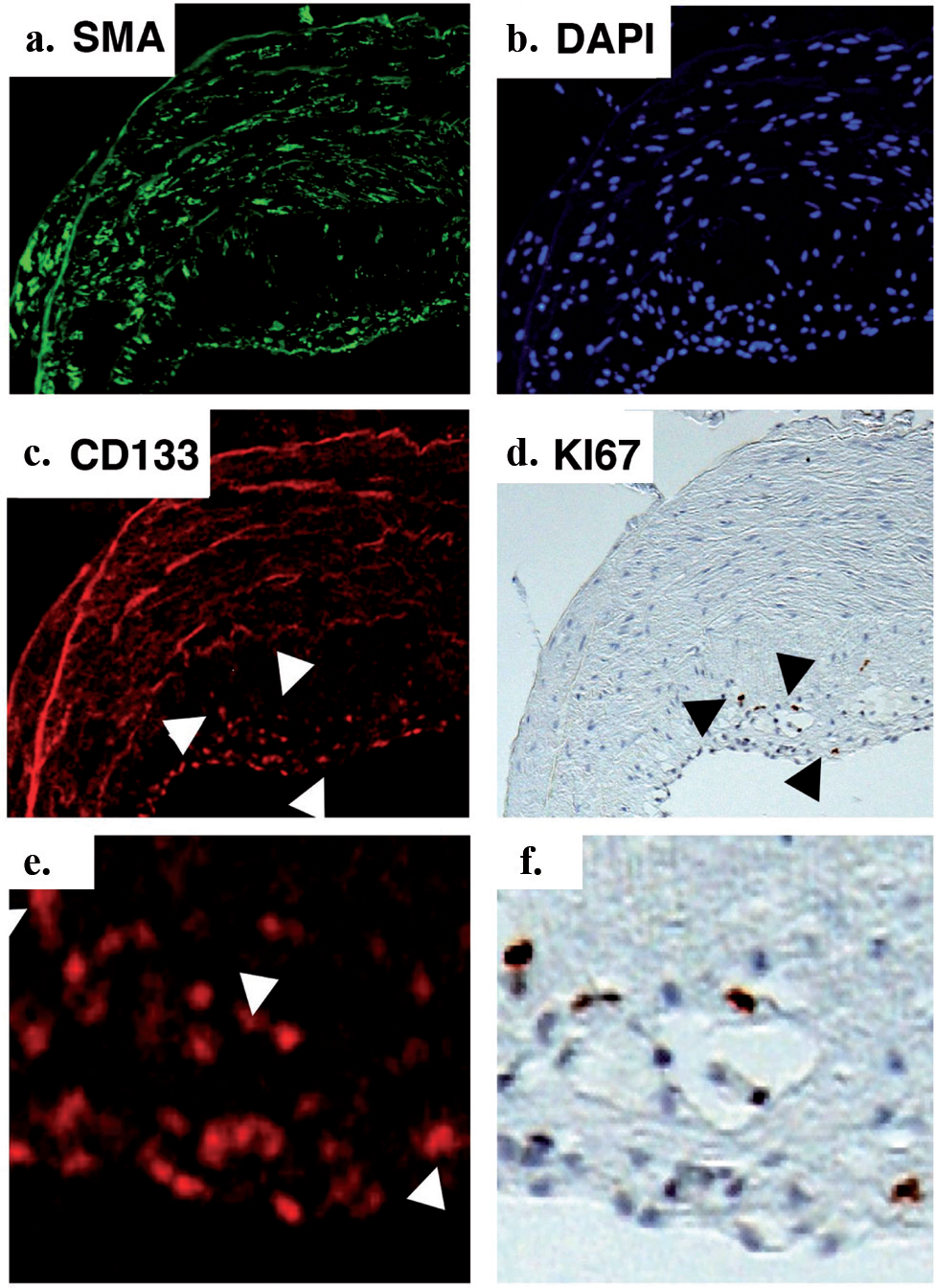
CD133 positive cells accumulate and proliferate in the pulmonary artery intima of PAH patients. Immunostaining with antibodies to smooth muscle actin (SMA) demonstrates the accumulation of mesenchymal cells in the intima (a) DAPI staining demonstrates total cell accumulation (b) CD133+ cells comprise a portion of the cells accumulating in the intima (c) Ki67 immunostaining on the same section, re-photographed, demonstrate that the majority of proliferating cells are CD133+ (d) High magnification views of (c) and (d) are shown in panels (e) and (f), respectively
A recent study has shown significant accumulation of c-Kit+ cells in the pulmonary arterial lesions of IPAH patients in an adventitial perivascular distribution.[127] These cells comprised both a mast cell and a potential progenitor population of cells. A marked expansion of the vasa vasorum was noted, with c-Kit+ cells present in this expanding vasculature. The findings were very similar to those reported by Davie et al. and supported the notion raised in that study that c-Kit+ and vasa vasorum could be therapeutic targets in PH.
Chemokine signaling involved in progenitor cell recruitment
At present, the molecular mechanisms controlling the recruitment, retention, and differentiation of circulating progenitor cells into the vessel wall remain largely undefined. It is clear, however, that chemokines play an important role in the vascular remodeling process, through the guidance of circulating mononuclear cells to the injury site and activation of resident vascular cells.[128,129] Considering the cell type-specific expression of chemokine receptors and the substantial overlap in ligand-receptor specificity, an interactive network of chemokines and chemokine receptors has emerged, which exhibits enormous plasticity in different types of injury / repair processes. A growing number of chemokine / chemokine receptor pairs with confined effects in vascular diseases have been described.[130] For instance, in systemic circulation, SDF-1 has been shown to regulate vascular repair by CXCR4-dependent SM-progenitor cell recruitment, which contributes to the maladaptive response to injury.[131] Furthermore, three distinct chemokine / chemokine receptor pairs (MCP-1 / CCR2, RANTES / CCR5, and Fractalkine / CX3CR1) have been shown to direct lesional leukocyte infiltration.[132] In addition, MCP-1 / CCR2 and Fractalkine / CX3CR1 increase expansion of neointimal SM-like cells.[133] Thus, to ultimately pharmacologically modulate the maladaptative responses in arterial remodeling, it will be essential to identify specific chemokine / chemokine receptor pairs that play specific roles in the remodeling process. It is possible that these will be vessel- and disease-specific and may, in fact, vary over time.
Recent studies have begun to address specific chemokine / chemokine receptor pairs involved in inflammatory / progenitor cell recruitment to the pulmonary arteries in models of pulmonary hypertension. Advancement in this area has come about through the use of laser capture microdissection (LCM), to evaluate vessel-specific changes, as it is becoming increasingly clear that an analysis of the whole lung tissue may not accurately reflect the specific changes in gene and protein expression in the vessel wall itself.[134–136] Of late, with the use of the LCM technique, sustained hypoxia-induced upregulation of a wide range of inflammatory mediators, growth-, differentiation-, adhesion- and fibrosis-associated molecules in a pulmonary artery-specific fashion [137] was demonstrated. The study demonstrated the upregulation of a number of chemokines and receptors, as well as adhesion molecules, which preceded the influx of the inflammatory cells and the onset of perivascular fibrosis. These included CXCL12 (SDF-1) / CXCR4, VCAM-1, ICAM-1, TGF-β, and 5-LO. Interestingly, over time, the pulmonary artery inflammatory microenvironment became more complex, with the appearance of other cytokine, adhesion, growth and differentiation mediators (such as IL-6, MCP-1, PDGF-BB), which was consistent with the increasing number and persistent presence of monocytes and other inflammatory cells in the perivascular regions.
Upon removal of the hypoxic stimulus, expression of specific chemokine / chemokine receptors and adhesion molecules rapidly returned to control levels, which preceded the disappearance of inflammatory cells and ultimately reversal of vascular remodeling and resolution of pulmonary hypertension. Importantly, administration of antagonists of SDF-1 / CXCL-12 and CXCR4 has been shown to prevent vascular remodeling and accumulation of c-kit+ / Sca-1+ progenitor cells in the vessel wall of chronically hypoxic mice.[138,139] Additionally, inhibition of SDF-1 / CXCR4 signaling reversed the established remodeling in these mice. These studies highlight the impact of progenitor cells in the pulmonary hypertensive process and hint at their potential therapeutic importance for pulmonary hypertension. Similarly, in patients with idiopathic PAH, increases in fractalkine, RANTES, and PDGF expression, particularly within the pulmonary arterial wall, have been observed.[136,140,141] Further studies are essential to define more precisely the chemokine / chemokine receptor pairs involved in the pulmonary vascular remodeling process. This information will be critical to the development of new therapeutic interventions.
SUMMARY
In summary, substantial experimental evidence points to the role of the circulating progenitor cells in vascular pathology, which characterizes chronic PH. More study is needed to determine the types of progenitor cells involved in vascular remodeling and their specific functions, once they take up residence in the vessel wall. Much study is needed to determine the factors involved in their recruitment and retention. Additionally, studies are needed to determine factors involved in controlling their proliferative and differentiation states. Studies are also needed to address the role of resident lung and vascular progenitor cells in the remodeling process. Collectively, these experiments may yield opportunities for critical new treatment strategies.