
Abstract
Developing new treatments for pulmonary arterial hypertension (PAH) is a challenge. We have enjoyed success with regulatory approvals for three drug classes—prostanoids, endothelin receptor antagonists and phosphodiesterase type 5 inhibitors. But we have also seen some disappointing results, for example, from studies with vasoactive intestinal polypeptide, statins and tergolide. Animal models are an unreliable predictor of efficacy in humans. The best model for the disease is the patient. This review discusses three major issues facing the evaluation of drugs in PAH patients—target validation, choosing the right dose, and early trial design.
Drug development is an expensive and risky business in any disease domain. The chances of a drug entering phase I trials reaching the market place has fallen to 5%.[1] The most common cause, accounting for around 50%, of failure of a drug in early clinical trials is lack of efficacy.[2] Safety is also an important concern, but accounts for less than one-fifth of withdrawals from early clinical development.
The drugs currently used to treat pulmonary arterial hypertension (PAH)—prostanoids, endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors—were originally developed for other conditions. The introduction of these drugs has not been without impact, as patients feel better and may live longer on these treatments than they did before. That said, it is arguable whether the current treatments affect the vascular pathology itself. A popular view is that these drugs may be working more to support and preserve the function of the right ventricle (RV) than reverse the structural changes in the pulmonary vascular bed. Clearly there is still more to do in this field.
The availability of medicines to treat PAH has stimulated considerable interest in the pathology of PAH and finding new drugs (Table 1). This has led to a long list of possible drug targets. A number of challenges impact on drug development in PAH, but key among them are (1) validating novel drug targets, (2) selecting the most appropriate dose of a novel agent, and (3) conducting informative clinical studies in a small patient population.
Novel agents for pulmonary hypertension
VALIDATING NOVEL DRUG TARGETS
The decision to take a new molecule into humans to manipulate pharmacologically a novel drug target—that is, to do the human experiment—depends on confidence in the drug target as well as safety considerations around the drug itself. The risks are less for a validated target and a re-purposed drug than for a novel target and a new chemical entity (Fig. 1).
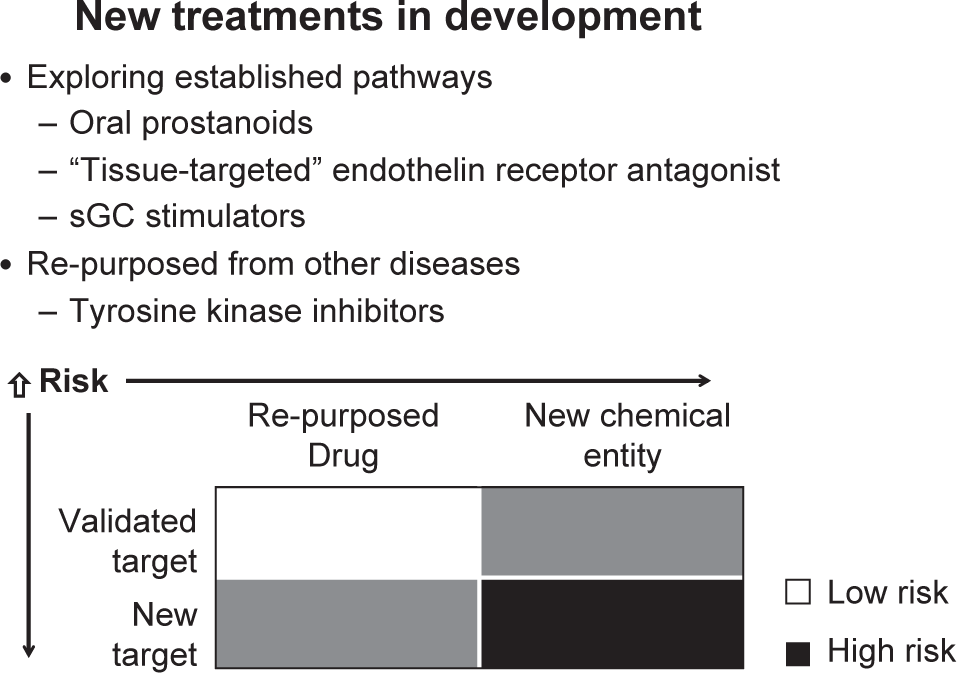
Risk evaluation for new treatments in development
Target expression
Clearly, evidence of expression of the drug target, for example a receptor, enzyme, ion channel, transcription factor etc – in the diseased tissue is important when deciding on its role in the disease. Expression of the target may be altered in a manner that is consistent with the known biology. Altered expression may be established by measuring gene transcripts, but protein levels are more convincing. Absence of a change in protein expression does not exclude that protein from a role in the disease, as phosphorylation is a major mechanism for altering activity without the need for altered levels. Conversely, evidence of altered expression of the candidate protein is supportive of a role in the disease but is not proof of causation. The problem with probing explanted end-stage lung tissue is that it is akin to visiting the scene of a crime. There are plenty of suspects hiding the true culprit, who may already have left the scene.[3]
Augmenting or blocking activity of the candidate molecule in animal models can be used to provide further support for a given target. There has been much discussion around the value of these models.[4] The literature is replete with pharmacological agents that work in animals, but not in humans.
Genetics
There is no doubt that the best model for human disease is the human, and a particularly powerful tool is genetics. Identifying a gene variant associated with PAH identifies a candidate pathway with a “motive” and a means of stratifying patients by genotype, but there are challenges even here. It is now over 10 years since the discovery of mutations in genes encoding bone morphogenetic protein receptor 2 (BMPR2), ALK1, and endoglin indicating a role for the TGF signaling pathway in PAH.[5,6] Patients with BMPR2 mutations appear to have a more aggressive form of the disease, presenting earlier, and dying younger.[7] There is no consensus on whether some mutations are more deleterious (in terms of worse prognosis) than others.[8,9] but it does seem likely that clinical trials that enroll patients with severe disease will enrich for patients with BMPR2 mutations and this needs to be understood when interpreting outcome data.
Dysfunctional mutations in BMPR2 are found in the majority of patients with hereditable PAH and around 20% of “sporadic” cases. This has focused attention on strategies for repairing BMPR2 signaling as a treatment for PAH, for example, by facilitating BMPR2 trafficking to the cell membrane or restoring the balance in BMP/TGFβ1 signaling by inhibiting the ALK5 receptor.[10] These approaches are attractive as they address fundamental molecular mechanisms underlying the disease but are currently experimental. Furthermore, as patients frequently present in an advanced stage of the disease, the extent to which targeting an early event will restore a normal pulmonary circulation is open to question.
Several other gene variants have been associated with PAH and as gene sequencing becomes more accessible, this number will increase. Investigators studying systemic diseases are looking at Mendelian randomization to provide insight into target validation and even safety assessment.[11] The numbers of patients required for this exercise likely limit the usefulness of this approach in PAH, but there may still be merit in pursuing extreme phenotypes.
Circulating biomarkers
An increasing number of circulating or soluble biomarkers have been put forward as candidates for predicting severity or survival and monitoring the course of PAH. To date, only BNP is used clinically in decision making, but a range of markers reporting on endothelial function, inflammation, coagulation, and oxidative stress have been suggested.
These circulating molecules tell us something about the disease, particularly its heterogeneity. An interesting observation is the range of values obtained from any PAH cohort for any given marker. Take BNP. The range of values for this circulating peptide range 100-fold, despite ‘standardisation’ for functional class, and embrace the normal range. Better understood, these markers represent intermediate phenotypes that can be exploited for drug target identification and as signatures that define patient groups that might respond to particular therapies.
For a circulating biomarker to be valuable in this respect, it has to inform on a relevant pathogenic pathway. An example of this is iron. Studies from our lab and others strongly suggest that iron deficiency without anemia is a marker of poor outcome in PAH.[12] Around 60% of patients had evidence of iron deficiency, which could not be explained by markers of inflammation, and this was linked with higher mortality. It is the subject of discussion whether iron deficiency is directly linked to pulmonary vascular disease, right ventricular function, or more peripheral actions on skeletal muscle that might affect exercise function. But iron deficiency is easily recognized and correctable, and we have embarked on a study where patients with iron deficiency are randomized to a single infusion of ferric carboxymaltose or saline, and pulmonary hemodynamics and exercise capacity are studied at two weeks and three months, respectively (ClinicalTrials.gov identifier NCT01447628). A similar biomarker-driven approach, based on, for example, levels of certain circulating cytokines, might be used to select patients suitable for anti-inflammatory drugs. Arguably, the patients who might benefit most are those with highest circulating levels of endogenous ligands.
FINDING THE RIGHT DOSE
There are three important questions to ask when administering a drug into humans for the first time: (1) does the drug reach the target; (2) does it modify activity through the target; and (3) what is the dose-response relationship? Understanding the answer to these questions enables the investigator to plan further clinical trials or conclude why a drug is ineffective.
Deciding on a suitable dose range for clinical trials is relatively straightforward for a drug that executes it therapeutic action by modifying vasomotor tone. This can be established by exploring the acute response of the cardiopulmonary circulation to the drug in the cardiac catheter laboratory. It is more challenging to identify a suitable dose for drugs directed at reversing pulmonary vascular remodeling.
Since the end game is to reduce pulmonary vascular resistance and improve cardiac output, one approach might be to employ novel devices for chronic ambulatory monitoring, as currently under trial in heart failure.[13] This is an evolving rather than an established and validated technology, but its appeal in PAH is that it may permit dose adjustment according to response, reducing the number of patients required in studies and enable early decisions on drug efficacy. It may also allow patients to participate in more than one clinical trial, such that the response to altering the dose of one drug at a time can be measured. It would also allow assessment of the effects of drug withdrawal and therefore the value of combination treatment.
Molecular imaging may also have value in the early evaluation of drugs in humans. Positron emission tomography (PET) is a noninvasive tool that is proving very useful in oncology. It may be used to investigate drug distribution, receptor occupancy, and biochemical response. It can provide proof-of-mechanism, an early readout of efficacy, and dose-response relationships.
18F-fluorodeoxyglucose (18F-FDG) PET is a useful paradigm. 18F-FDG has been used to study adaptation of the myocardium to pressure-load, but it may also provide a useful signal from the lung parenchyma. Proliferating cells exhibit the Warburg effect, whereby increased glycolysis drives increased glucose uptake. This phenomenon has been demonstrated in pulmonary artery endothelial cells in culture, and a report of increased FDG uptake in the lungs of patients with PAH is consistent with this.[14] Animal studies suggest that the lung signal can be reduced by antimetabolic and antiproliferative treatments that reduce pulmonary hypertension in animal models.[15] If reproduced in human studies, FDG PET may provide a bridging biomarker for exploring dose-response relationships for antiproliferative/antimetabolic drugs in PAH patients.
NOVEL TRIAL DESIGNS
PAH is not a common disease. An important obstacle to drug development, as is the case with orphan diseases in general, is the ability to recruit a large enough sample of patients to draw inferences about a new drug's efficacy and safety. For most accepted intermediate or ultimate end points, the prevalence of PAH is insufficient to support formal examination of all potential drug targets.
As a result, investigators are turning to adaptive trial designs, based on Bayesian statistics.[16] The development of Bayesian approaches to the design and analysis of studies in the exploratory phase of drug development has accelerated over the last decade.These approaches have utility in both traditional Phase I studies as well as in Phase IIb dose selection studies.
Adaptive dose-ranging trials are more efficient than traditional approaches. First, they are designed to address explicitly the goals of the individual trial through choice of appropriate decision criteria. Second, the approach to analysis in such designs recognizes that it is more appropriate to estimate the dose-response relationship than to compare individual dose to placebo. Finally, the design reduces the chance of having to rerun a phase IIb design by a poor choice of a few doses.
There is a tendency to design studies for drugs repurposed from other diseases around doses approved for the original disease indication. It is important that dose–response relationships are critically evaluated in PAH patients, particularly, as it may be possible to prescribe lower doses. An example would be sildenafil. It is also important to emphasize from the perspective of drug safety that drugs from the same class are not all viewed the same. While the tyrosine rector antagonist, imatinib, may have beneficial effects in some patients with PAH,[17] a recent report suggests dasatinib use in chronic myeloid leukaemia is associated with the development of the disease in some patients.[18]
CONCLUSIONS
We may have achieved as much benefit as we are likely to see using drugs with primarily a vasorelaxant effect. Future progress depends upon identifying drugs with novel modes of action. There are many attractive candidates, but not all will progress to late phase clinical trials. It would be helpful to the academic and pharmaceutical community to define a checklist that might aid candidate selection and reduce the risk of failure in human studies (Table 2). Confidence in the validity of the drug target is increased by genetic data associating the target with the disease. More practical at this time is a circulating biomarker linking the target with a mechanism of tissue damage. Both genetic and circulating biomarkers permit patient stratification in clinical studies. Novel technologies and trial designs offer useful approaches to dose selection and may allow early go/no-go decisions in patients with the disease.
Checklist for novel drugs and targets
