
Abstract
Despite recent advances in cancer diagnosis, prevention, detection, as well as management, the disease is expected to be the top cause of death globally. The chemotherapy approach for cancer has become more advanced in its design, yet no medication can cure enough against all types of cancer and its stage. Thus, this review aimed to summarize a recent development of new therapeutic agents and novel drug targets for the treatment of cancer. Several obstacles stand in the way of effective cancer treatment and drug development, including inaccessibility of tumor site by appropriate drug concentration, debilitating untoward effects caused by non-selective tissue distribution of chemotherapeutic agents, and occurrence of drug resistance, which leads to cross-resistance to a variety of drugs. Resistance to treatment with anticancer drugs results from multiple factors and the most common reason for acquiring drug resistance is marking and expelling drugs that prevent cancer cells to be targeted by chemotherapeutic agents. Moreover, insensitivity to drug-induced apoptosis, alteration, and mutation of drug target and interference/change of DNA replication are other main causes of treatment failure.
Introduction
Cancer
Cancer refers to a group of diseases characterized by uncontrolled, fast, and abnormal cell proliferation. 1 There are various types of cancer depending on the origin and organ of the body, which they affected, but they have common features: abnormal cell growth, capacity to invade other tissues, and spread to another distant organ through a blood vessel and lymphatic systems. Although the specific etiology of cancer is unknown, several chemical agents, genetic factors, nutrition, hormones, and medicines all play a role. 2 Cancer cases and deaths are on the inexorably rise over the world, despite the fact appropriate care is taken in current modern medicine. Researchers should focus on a new approach to cancer treatment involving the pathophysiology of the diseases, the discovery of human genome sequence, and novel molecular targets that kill cancer cells. 3
The management of cancer requires pharmacologic and non-pharmacologic treatment algorism like surgery, radiation, and chemotherapy, depending on the stage of the diseases, types of cancer cells, and patient’s clinical status. 4 Since most cancer is not curable in an advanced stage, prevention is the most important and active area of research. Both lifestyle modification and chemopreventive agents significantly reduce the risk of developing cancer.5,6
Epidemiology of cancer
The worldwide burden of cancer is rising as the world population ages and grows, as well as increased adoption of cancer-causing habits such as smoking, eating processed foods, and leading a sedentary lifestyle. 7 In the developed world, it is the second top cause of death. Worldwide cancer has increased to 18.1 million cases and 9.6 million cancer deaths, according to GLOBOCAN 2018 data. Underdeveloped countries accounted for 56% of cases and 64% of deaths, respectively. 8 Overall mortality due to cancer is nearly the same, although the incidence of cancer in developed countries is half of the developing countries. 9 Despite the increased incidence and prevalence, cancer remains a low priority for public health in Africa due to inadequate economic resources and other communicable and non-communicable diseases. 10
Cancer is responsible for roughly 6.2% of all deaths in Ethiopia, and it is postulated that the annual incidence of cancer is around 68,960 cases and the annual death is over 54,000. Breast cancer, cervix cancer, and colorectal cancer are the three most common cancers in Ethiopia’s adult population. 11
Pathophysiology of cancer
The mechanism of cancer pathogenesis is still a mystery and it is not completely understood. Normal cell growth and proliferation control systems are thought to be disrupted. 12 Normal cells would have cancer in a stepwise approach and the first event to occur is initiation. It occurs when a carcinogenic substance encounters normal cells to produce genetic damage and result in the mutated cell. Promotion of the environment is altered by carcinogens or mutated cells over the normal cell growth. 13 Third, transformation (conversion) occurs when the mutated cell becomes malignant, between carcinogenic phases and clinically observable tumor development, up to 20 years may elapse depending on the kind of cancer. Finally, progression occurs when cell proliferation takes over and the tumor spreads called metastases.10,14
Oncogenes and tumor suppressor genes are the major genes involved in cancer. Proto-oncogenes are normal genes that present all normal healthy cells that regulate cell function and replication. Genetic damage of proto-oncogene causes point mutation, chromosomal rearrangement, or increased gene function, resulting in an oncogene. Oncogene produces abnormal or excessive gene product that disrupts normal cell growth and proliferation and it becomes cancerous. 15
Tumor suppressor genes prevent abnormal cellular development and proliferation. P53 is one of the most prevalent tumor suppressor genes, with p53 mutations accounting for up to 50% of all cancers. When p53 is deactivated, the cell permits mutations to occur, which leads to cancer. 16 Other genes and signaling which are involved in the pathogenesis of cancer include Bcl-2 (B cell lymphoma-2), A stem cell factor (SCF), and cancer stem cells (CSCs).17–19
Clinical presentation and diagnosis method
Cancer patients may exhibit a variety of signs and symptoms. Many people are afraid of being diagnosed with cancer, so they may not seek medical help when the symptoms first appear when the disease is most treatable. After the initial visit with a clinician, a variety of tests will be performed depending on the initial differential diagnosis. 10
An appropriate laboratory test, radiologic scans, and tissue samples are necessary. The sample is taken from the tissue by using a method like biopsy, fine-needle aspiration, or exfoliates cytology. No treatment should be initiated without a pathologic diagnosis of cancer. Depending on the type of cancer, genetic analysis provide additional information on the prognosis of the malignancy and whether a certain therapy may be appropriate. 10 Once the pathology of cancer is established, the staging of the disease is done before treatment is initiated. The TNM method, which specifies the tumor (T), nodes (N), and metastases (M), is the most often used leveling approach for solid tumors (M). Each letter is given a numerical value to represent the severity or extent of the ailment.10,20
The staging of cancer is done according to the primary tumor size, the extent of lymph node involvement, the presence of metastasis, and is usually referred to as stages I, II, III, or IV. Not all cancers can be staged according to this system, but many of the solid tumors are.10,21 Staging of the diseases is an important part of determining the prognosis of cancer and is used as a guide for the initial and further treatment of cancer. 22
Risk factors for cancer
Normal healthy cells are strictly regulated with stimulatory and inhibitory signals in a delicate balance. 23 When normal cells are subjected to physical, chemical, or biological stress, genetic changes occur, which proliferate and disrupt normal cell division, resulting in unrestricted growth, invasion, and metastasis. 24 Although the exact cause is unknown, chemicals (e.g. tobacco, benzene, aniline), environmental factors (e.g. ultraviolet radiation), genetics, viruses (e.g. human papillomavirus (HPV) and Epstein–Barr virus (EBV)), drugs, and diets are some of the suggested triggers. 7
Managements of cancer
The treatment goals are determined by the cancer stage and other conditions affecting the patient. A patient can receive chemotherapy to reduce disease progression, alleviate signs and symptoms, and cure the disease if possible.9,10
Pharmacologic management of cancer
Currently, surgery, radiation therapy (RT), and chemotherapy are the principal treatment strategies against cancer.25–27 Chemotherapeutic agents typically have a very narrow therapeutic index. 26 The dose should be adjusted based on body surface area. If too much is administered, the patient may suffer from fatal toxicities and too little is given, the desired effect on cancer may not be achieved. Many chemotherapy drugs have severe organ toxicity, making it impossible to treat cancer with progressively increasing doses. The dose of chemotherapy must be given at the frequency that allows the patient to recover from the toxicities of chemotherapy, each period of chemotherapy dosing referred to a cycle.10,28,29
Dose density chemotherapy is a shortening of the period between the cycles of chemotherapy and the number of required cycles in a short period. The regimen often requires the use of colony-stimulating factors (e.g. granulocyte colony-stimulating factor or Filgrastim) or to be administered to shorten the duration and severity of neutropenia.30,31
The underlying principles of using combination are agents with different pharmacologic actions, different organ toxicities, are effective against the tumor and ideally synergistic when used together, and that does not result in significant drug interactions.32,33 The conventional anticancer drugs are summarized in Table 1.
The overview of conventional anticancer drugs.
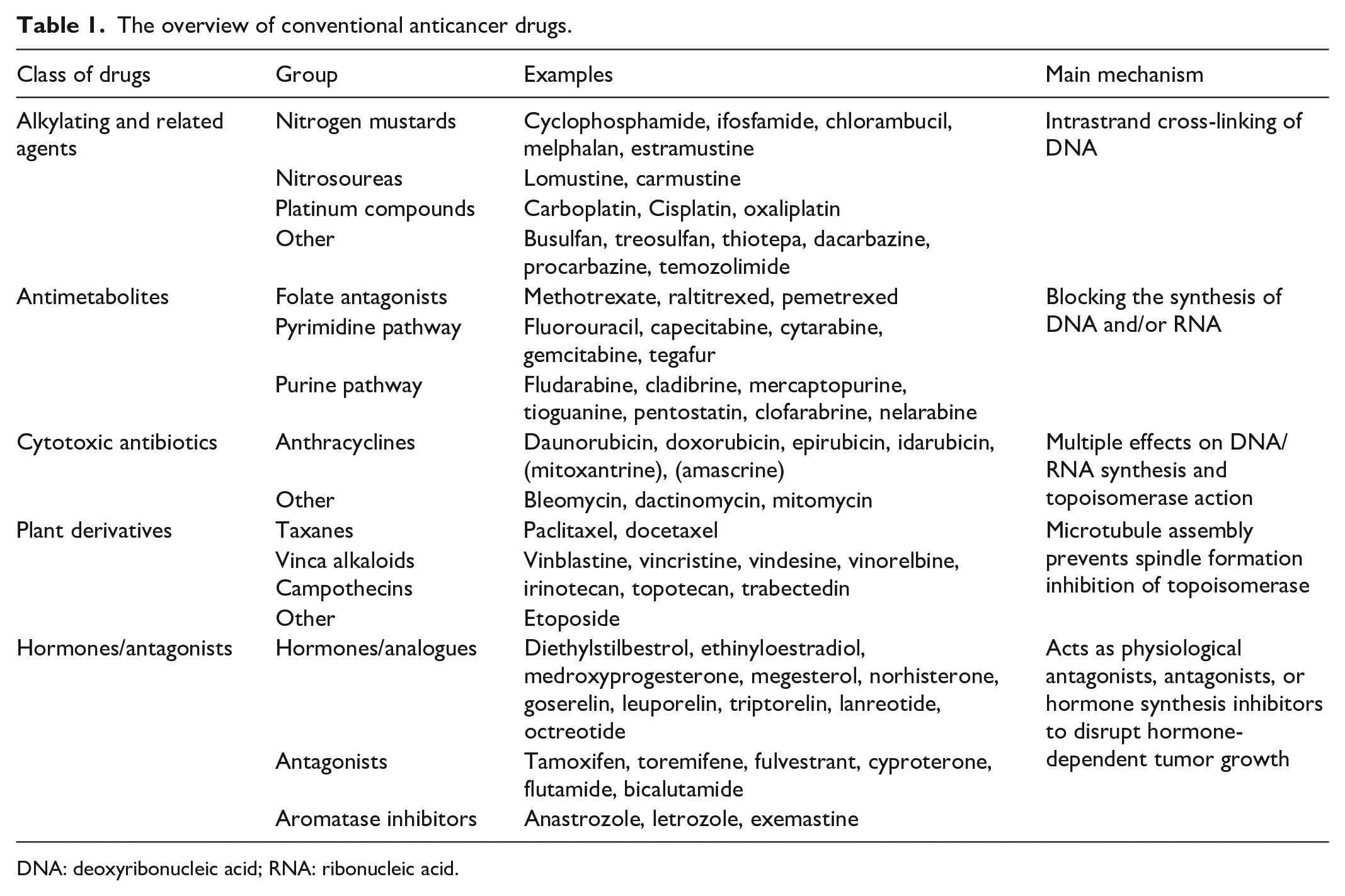
DNA: deoxyribonucleic acid; RNA: ribonucleic acid.
Non-pharmacological management of cancer
Pain is a common symptom experienced by a patient with advanced cancer; the treatment of such pain is often suboptimal. To manage it, non-pharmacological interventions are recommended, such as hot/cold therapy, physical therapy, and diet. Surgery and radiation are the main non-pharmacological modalities of therapy.34,35
New therapeutic agents and novel drug target
Because the human genome has been sequenced and genetic technology has advanced, there is a growing body of knowledge on cancer’s genetic changes, initiation and proliferation, therapeutic mechanisms, and novel treatment targets.36,37 Understanding the pathophysiology of the disease, human gene sequence, and the discovery of novel molecular targets is the core of modern medicine to conquer cancer therapy. Numerous noteworthy advances have been made in the development of targeted therapies. These targeted therapies are designed to attack cancer cells while demonstrating less damage to normal healthy cells. 21
Target therapies are drugs or other substances that block the growth and spread of cancer by interfering with specific molecules or targets that are involved in the growth, spread, and progression of cancer. Targeted therapies are currently the center of much anticancer drug development; hence, they are the cornerstone of precision medicine.38,39 The new therapeutic agents and novel drug targets for cancer treatment are summarized in Table 2.
Summary of new therapeutic agents and novel drug target for cancer treatment.

NSCLC: non-small-cell lung cancer; NTRK: Neutrophilic tyrosine receptor kinase; EZH2: Enhancer of zeste homology 2; ALK: anaplastic lymphoma kinase; EGFR: epithelial growth factor receptor; HER2: Human epidermal growth factor receptor 2; FLT3: Fms-like tyrosine kinase 3; PARP: poly (ADP-ribose) polymerase; FGFR: Fibroblast growth factor receptor; DNA: deoxyribonucleic acid.
Angiogenesis inhibitors
Angiogenesis is the way by which cancers cells develop blood supply for growth. It is crucial particularly as tumors become enlarged. 40 Many anti and pro-angiogenic factors play a role in the development of blood vessels. During blood vessels formation, different angiogenic factors are released and will initiate the proliferation, migration, and invasion of endothelial cells in new vascular structures. These factors are fibroblast growth factor (FGF), transforming growth factor-β (TGF-β), angiogenin, and vascular endothelial growth factor (VEGF).10,41–43
Angiogenic inhibitors are a novel class of drugs zeroing to disrupt tumor vascularization. In tumor progression, invasion, and metastases VEGF-A is over-expressed. Currently, VEGF-A and VEGFR2 inhibitors are available which target VEGF-A.9,44 Four main strategies are used in anti-angiogenic agents: the identification and application of natural angiogenesis inhibitor, the inhibition of endogenous factors that promote the formation of blood vessels, incapacitation of actively proliferating endothelial cells, and the inhibition of molecules that promote the invasion of the surrounding through the tumor blood vessels. Such agents do have low systemic toxicity and side effects and they do inhibit vascular growth factor signal transduction, reduce the tendency to induce resistance, boost anti-angiogenesis pathways, and display tumor angiogenesis specificity.10,45 Angiogenesis inhibitors including bevacizumab and ramucirumab are used for non-small-cell lung cancer (NSCLC). These drugs target VEGFs.45,46
DNA synthesis inhibitors
Mutagenesis occurs when DNA is synthesized far more than normal. Identification and development of anti-folates as therapeutic agents need the understanding of folate derivatives. 47 Via dihydrofolate reductase (DHFR) cells produce polyamines, thymidylate, and purines. Serum folate deficiency among patients with acute leukemia leads to the new postulation that acute leukemia might be the result of folate deficiency. Ribonucleotide reductase (RNR) is an enzyme needed for the de novo conversion of ribonucleotide diphosphate (NDP) to deoxyribonucleoside diphosphate and helps to regulate the supply of intracellular dNTP. Imbalance substrates for DNA synthesis lead to mutagenesis and cell death. 48
Drugs targeting folic acid receptor
Folic acid receptor (FAR) being a known tumor marker and the chemistry to therapeutic and imaging agents, folic acid is a clinical tumor-targeting ligand in DDS. 49 Among four folate receptors (FARα, FARβ, FARγ, and FARδ), two of them, namely, FAR-α and FARβ are overexpressed in different types of cancers, that is, colon, bladder, ovarian, lung, breast, pancreas, kidney, and prostate cancer. 11
Tumors expressed 25% and 40% of FARα and FARβ, respectively. Moreover, in inactivated myeloid cells of the immune system, FARβ is majorly expressed. FA is thus a promising molecular target for cancer and inflammatory/auto-immune illnesses. Drugs targeting this site include Hydroxyurea: Produces inhibition of DNA synthesis and induction of cell death through blocking the synthesis of deoxy-nucleotides by inactivating ribonucleoside; Cytosine-β D-arabino-furanoside is another selective inhibitor of DNA synthesis. 50 A humanized monoclonal antibody against such as Farletuzumab (MORAb-003) also showed a promise both in vivo and in vitro studies against FARα. 11
Transcription regulators
Living cells need transcription for survival and growth. Although, tumor cells need high levels of transcription such as mRNA and ribosomal RNA transcription through RNA polymerase II and RNA polymerase I, respectively. DNA transcription highly depends on the interaction between transcription regulatory components and transcriptional machinery. 11
Several transcription factors such as aberrant activation, repression, and temporal/spatial discoordination, causing transcription deregulation with structural variations such as translocations, transcriptional, fusion, and mutations processes lead to cancer initiation. 10
Novel immune therapies are developed against some transcription factors. Different therapeutic agents can target transcription machinery such as components of associated transcriptional complexes, RNA polymerases, and cyclic-dependent kinases. Transcription factors that control angiogenesis, growth, invasion, metastasis-related genes, and apoptosis in tumor cells are possible molecular targets for cancer gene therapy. Homoharringtonine (transcription regulator) binds to the 80S ribosome in eukaryotic cells and constrains protein synthesis by affecting the chain elongation. 10
Enzyme inhibitors
Enzymes are a potential therapeutic target and their embarrassment is accountable for facilitating apoptotic death in cancer cells. Inhibitors of metabolic enzymes in cancer therapy regimens enhance the efficacy of chemo/radiotherapy. 9
Estrogens (ER) affect breast cancer by causing genetic mutations and/or affecting DNA repair processes. Aromatase, a CYP450 enzyme, is encoded by the aromatase gene CYP19 and is involved in the synthesis of estrogens.9,51 Aromatase is found in all body tissues such as the brain, bone, skin, muscles, and breast. Inhibition of this enzyme is accountable for the decrement of estrogen level. Therefore, in the development and progression of hormone-responsive breast cancers, aromatase enzyme may have a substantial role and their inhibitors can be used as chemopreventive therapeutic agents. 7
Protein kinase C
Protein kinase C (PKC) plays a key role in the arrangement of the differentiation/proliferation, cell division, and cell cycle. The mechanism of expressions and activation of PKC is affected in different kinds of cancer. PKC fits into the threonine/serine kinase enzyme family. Based on their biochemical properties, structure, and domain, the isozymes of PKC are divided into three classes. Conventional PKCs consists of α, β1, β2, and classical isoenzymes. Conventional PKCs are a target of phorbol esters that promote tumor formation. 52 Many PKC isoforms have varied functional features due to cellular and intracellular localization differences, and each isoform has its signal sensitivity. 11
The attendance of diverse activation and tissue distribution PKC isoforms exhibited the need to design inhibitors specific to PKC isoenzymes. PKC isoenzyme activated by phorbol esters that activate the tumor formation process has a significant role in cancer management. 21
Results indicated superior anticancer and anti-estrogenic action of Z-endoxifen over tamoxifen in the AI-sensitive and resistance settings, using the letrozole-sensitive MCF7 aromatase expressing (MCF7AC1) cell line and its matching letrozole-resistant (MCF7LR) cell line. Follow-up studies demonstrated that Z-endoxifen, but not tamoxifen, binds to protein kinase C beta one (PKCβ1), a member of the serine/threonine-specific protein kinase family that control signaling pathways intricate in tumorigenic transformation and cell proliferation. Apart from these, PKCβ1 silence similar to Z-endoxifen, potently inhibits ER+ cell proliferation, and additionally causes ERα turnover—the potential mechanism by which PKCβ1 may mediate anti-proliferative effects in ER+ breast cancer cells. 7
DNA intercalator and groove binding agents
Groove binding and intercalation are the key apparatuses underlying DNA–drug interaction. Function and recognition of DNA-associated proteins (transcription factors, topoisomerases, DNA repair systems, and polymerases) are affected by DNA intercalating agents. Groove binders and intercalators have confirmed the clinical usefulness of anticancer agents due to selectivity. 11
The intercalators can be non-toxic or toxic reliant on the absence/presence of numerous functional groups: cationic, electrophilic, or basic required for Genotoxicity. Groove binding molecules do not induce large conformational changes in DNA and unlike intercalators bind to the minor groove of DNA as a standard lock-and-key model. 10
Groove binding molecules are more sequence selective and do not display G-C region preference, whereas DNA intercalators are less sequence selective and display a preference for G-C regions. Intercalators and groove binders have confirmed clinical anticancer agents. Mitomycin and anthracyclines are both DNA cross-linker or groove-binding molecules.10,11
Drug targeting tubulin (mitosis)
A chemotherapeutic agent that Target tubulin cause cell-cycle arrest and apoptosis. The operation of the mitotic spindle agents that change microtubule dynamics has demonstrated broad-spectrum activity in the treatment of cancer important target for new cancer chemotherapeutic development. β-tubulin has distinct binding sites identified for taxanes, colchicine, and vinca alkaloids. 53
Epothilones are chemically distinct and the key benefits over taxanes include enhanced aqueous solubility, which eliminates the need for Cremophor-based formulations that can induce an immune response, decreased likelihood of P-gp-based resistance, and efficacy against taxane-resistant tumors. 9 A total of four epothilones have progressed to advanced clinical testing in different trials, including epothilone B (patupilone) from Novartis, analogues of epothilone B from (ixabepilone or BMS-247550) and Schering AG (ZK-EPO or ZK219477), and epothilone D (KOS-862) under co-development.53,54
Drugs targeting lectins
Lectins are non-immunological proteins with a preference for recognizing and binding specific carbohydrates linked to glycoproteins. To target cancer cells, carbohydrate–lectin interaction has been established. Lectins are required for biological processes including endothelium adherence, tumor vascularization, extracellular matrix adhesion, and tumor cell survival. 10 Some lectins of plant and animal origin persuade autophagy and apoptosis of cancer cells and therefore possess the potential of being advanced into anticancer medications. Lectins including mistletoe lectin, C-type lectins, annexins, galectins, concanavalin A, and galectins have revealed apoptotic inducing properties via several mechanisms. 55
Neutrophilic tyrosine receptor kinase gene change
TRK is a family of high-affinity cell membrane receptors with analogous intracellular signaling pathways and structures but distinct modes of stimulation and control. As a therapeutic target, this receptor binds to certain growth factors and is involved in various key activities for cell activation and survival, including death, differentiation, and growth. 56 Neutrophilic tyrosine receptor kinase (NTRK) comprising a family of genes such as NTRK-1, NTRK-2, and NTRK-3 found on chromosomes 1(1q22), 9 (9q22), and 15(15q22) and encoding for the TKR-A, TKR-B, and TKR-C proteins, respectively. They were recognized as high-affinity neurotrophin receptors then described and identified as oncogenes in colorectal cancer.9,10
Oncogenic activation of NTRK occurs in numerous ways like structural chromosomal rearrangements leading to splice variants, gene fusions, copy number alterations, mutations, and increase alteration and expression. 11 Generally, NTRK fusions are considered a path-genomic marker of specific neoplasms such as mammary analogue secretory carcinoma of the salivary glands, breast secretory carcinomas, infantile/congenital mesoblastic nephroma, and infantile fibrosarcomas. Larotrectinib and entrectinib are targeted drugs that stop the protein made by the normal NTRK genes. 56
Enhancer of zeste homology 2 (EZH2)
EZH2 is a potential target for cancer therapy and is expressed in a different type of cancer. EZH2 acts as a histone methyltransferase mostly via its SET domain, and it can co-activate or suppress transcription. EZH2 is a member of a family of polycomb group genes, which is a group of important epigenetic supervisors that suppress transcription. The oncologic role of EZH2 is to regulate cell cycle progression; dysregulation of EZH2 accelerates cell prolonging cell survival and proliferation, which results in cancer. 52
Ectopic expression of EZH2 in breast epithelial cells may persuade promotion of malignant cell division and growth, which is the demonstration of the oncogenic role of EZH2 to also obstruct apoptosis by regulation of E2F1-dependent apoptosis via modulating expression. 52 Many novel drugs targeting EZH2 have been researched, but only tazemetostate was approved. Great promising development of EZH2 inhibitors with high selectivity, low toxicity, and high efficiency is one of the upcoming approaches for cancer management. 57
Drugs acting on anaplastic lymphoma kinase gene change
Anaplastic lymphoma kinase (ALK) is typical for receptor-protein tyrosine kinases, which comprises three domains: a cytoplasmic tyrosine kinase domain, a single-trans membrane-spanning domain, and an extracellular ligand-binding domain, that plays a key role in cancer pathogenesis. 58 Approximately 5% of NSCLC have a rearrangement in a gene named ALK. This change is frequently understood in light smokers or non-smokers commonly on younger’s who have adenocarcinoma subtype of NSCLC. ALK gene rearrangement results in an anomalous ALK protein that can cause a cell to spread and grow. 10 The greatest rearrangement ALK rises from the inversion of the short arm of chromosome 2 that creates a fusion between the 3′ portion of ALK gene and the 5′ portions of EML4 gene. More than 7 ALK gene rearrangement variants have been designated relating to different non-EML4 fusion partners and EML4-ALK breakpoints. ALK rearrangement is frequently recognized in tumor tissue through reverse transcription-polymerase chain reaction. Although ALK gene rearrangement affects only 4% of all lung cancers, they are more common in adenocarcinomas, even with non-smokers, and seem almost conjointly exclusive with triggering KRAS or EGFR mutations. Drugs that target the abnormal ALK protein are lorlatinib and ceritinib.58,59
Drugs act on epithelial growth factor receptor gene changes
Epithelial growth factor receptor (EGFR) has been the most investigated of the four members of the epithelial growth factor receptor family (ErbB). 10 All convey the signal from the microenvironment into the cells and express in almost every cell. Each process in the cell such as differentiation and migration, survival, cell and tissue morphogenesis, and proliferation involves ErbB receptors signaling. Due to deletion mutations, point, and gene amplification, the ErbB signaling pathway is commonly originating too active in epithelial cancers, accounting for 80%–90% of all cancer cases. This alleyway is a promising target in the oncology field. 11 The two main tailor therapeutic strategies are one uses small tyrosine kinase inhibitor molecules that would inhibit the oncogenic form of the receptor and target the receptor’s kinase. The second option is to use monoclonal antibodies like bevacizumab, nivolumab, and cetuximab that target the extracellular ligand-binding domain of the receptor, therefore, terminating the signaling either via preventing its dimerization or increasing the rate of receptor degradation. 21
EGFC is a protein that is located on the surface of cells that aids cells to divide and grow, however, some NSCLC cells have too much EGFR, which makes them grow faster. Medications such as gefitinib and erlotinib are EGFR inhibitors that can hinder the signal EGFR that activates cells to grow fast. 60
Human epidermal growth factor receptor 2
Understanding the biology of breast cancer leads to the distinguishing of several potential therapeutic agents for can cancer management. Particularly the targeting properties of the immune system provide an approach to increase the selectivity of anticancer treatment. 61 Trastuzumab, the humanized anti-human epidermal growth factor receptor 2 (HER2) monoclonal antibody, which activates the immune system and targets HER2 to block the physiologic function of the HER2-signaling network has been revealed a noticeable therapeutic value in patients with HER2-positive metastases breast cancer.9,62 HER2 is overexpressed about 20% of all breast cancer. Trastuzumab and pertuzumab and antibody–drug conjugates (trastuzumab emtansine and trastuzumab druxtecan) have a clinical outcome for HER2-positive breast cancer. 21
Fms-like tyrosine kinase 3 as a therapeutic target in leukemia
The association of Fms-like tyrosine kinase 3 (FLT3) mutations with WBC counts at diagnosis and early death was investigated in acute promyelocytic leukemia patients. The documentation of recurring driver mutations in genes encoding tyrosine kinase leading to the development of molecular targeted treatment strategies designed to increase the outcome of acute myeloid leukemia patients. 63
The FLT3 is the most frequently mutated gene in AML, with internal tandem duplications within a juxta-membrane domain (FLT3-ITD) or missense mutation in the tyrosine kinase domain which accounts for 30%–35% of AML patients at diagnosis. 64 It has appeared as a potential therapeutic target and therefore encourages the synthesis of FLT3 tyrosine kinase inhibitors. Although the therapeutic benefit of FLT3 inhibition, predominantly as a monotherapy, commonly leads to disease relapse and treatment resistance. 21
Second-generation small molecule inhibitors of FLT3 such as gilteritinib, crenolanib, and quizartinib display more specific and potent FLT3 inhibition with high efficacy and less toxicity for the treatment of leukemia. 65
Enzyme poly (ADP-ribose) polymerase (PARP) inhibitors
PARP proteins are implicated in several cellular processes such as DNA damage repair, transcriptional regulation, and DNA replication. PARP proteins have been examined extensively, particularly PARP1 and PARP2, which are linked to DNA stability. The PARP enzyme is involved in the regulation of cellular responses to DNA damage and excision repair of single-stranded breaks. The base excision repair (BER) pathway removes and recognizes inappropriate or damaged bases. 46 This leads to the development of the potentially cytotoxic apyrimidinic or apurinic site. PARP binds to the strand break and relaxes the chromatin structure to permit easier entree to the BER machinery. 9
PARP transfers ADP- ribose unit from NAD+ to itself, histones, and nuclear target proteins. These forms branched and long polymerase of poly (ADP-ribose) on the PARP enzyme, which acts as a signaling mechanism to recruit the BER machinery, such as PCNA, ligase III, RFC, polymerase β, and adaptor factor XECC1. When PARP is repressed, common single-strand breaks are transformed into double-stranded breaks in the course of DNA replication. 10
Homologous recombination is the most public mechanism of repairing double-strand breaks particularly in normal cells that maintain genomic stability. In the existence of injurious BRCA genes, homologous recombination is impaired. These tumors and cells that they form amplified vulnerability to PARP inhibition, which results in decrement of chromosomal stability and eventually cell death. PARP enzyme inhibitors such as niraparib, rucaparib, and olaparib are commonly used for the management of ovarian and breast cancer. 46
Fibroblast growth factor receptor protein-tyrosine kinase inhibitors
Fibroblast growth factor receptors (FGFRs) are extremely conservative TKR that is central in the development of human cancer. 9 The family contains four extremely conserved TKR such as FGFR1, FGFR2, FGFR3, and FGFR4, containing three extracellular immunoglobulin type domains a cytoplasmic tyrosine kinase domain, a single transmembrane domain, and (D1-D3). 66 Gene augmentations are the most common FGFR alterations in human cancers accounting for 66% of all FGFR aberrations. FGFR1 is the most common amplified gene regularly detected in colon, lung, and breast cancer. FGFR gene alteration happens in different types of cancer such as the bladder, prostate, endometrium, breast, lungs, and stomach. Erdafitini is the FGFR antagonist effective for the treatment of such cancer. 21
Drugs targeting transferrin receptor
Transferrin is a plasma protein required to transport iron to proliferating cells. The transferrin receptor (TfR) is a cell-membrane-associated glycoprotein which involved in the regulation of cell growth and the cellular uptake of holo-Tf (iron-bound to Tf). Increased levels of TfR are predominantly expressed in ovarian, brain, colon, and lung cancer. Because of this, Tf is a vital ligand in the active targeting of tumors. TfR-targeted agents against several kinds of cancer display significant cytotoxic effects. For instance, saxenaet synthesized a drug delivery system (DDS) of a Tf-modified cytochrome C to target TfR on A549 lung cancer cells. This finding revealed that this conjugate design would be effective in cancers with increased expression of TfR because of its nontoxic pattern to lung normal cells. 10
B cell lymphoma-2
A defect in apoptosis is very important in cancer development and a major obstacle to effective treatment. BCL-2 family proteins regulate programmed cell death. Some members of this family (such as BAX and BAK) promote cell death, whereas other some members of the family (such as BCL-2 and BCL-XL) inhibit apoptosis. 67 Although the exact mechanism by which BCL-2 family regulates apoptosis is yet to be studied, it is thought that this family protein extends the survival of cell by inhibiting the programmed cell death in diverse tissues in response to a variety of stimuli through the regulation of cysteine proteases caspases. This mechanism is orchestrated without affecting cellular proliferation. 17 BAX and BAK also promote caspase activation through their effect on mitochondria. These two pro-apoptotic proteins induce the release of proteins from the space between the inner and outer mitochondrial membranes. This process results in the release of cytochrome c and other soluble proteins into the cytosol, which eventually leads to cell death. 68
The anti-apoptotic family members, such as BCL-2 and BCL-XL, inhibit BAX and BAK. Moreover, a study showed that BH3-only proteins disinhibit BAX and BAK by direct binding and inhibition of BCL-2 and another anti-apoptotic family member. Currently, there are numerous drugs that target the BCL-2 family which acts through potentiating its action or antagonizing it. 68
BH3 mimetics that target BCL-2
The BH3 mimetics are novel therapeutics that mimic the binding of BH3-only proteins to the hydrophobic groove of anti-apoptotic proteins. 69 The biological activity of BH3-mimetic compounds is dependent on BAX/BAK pro-apoptotic proteins and they are characterized by binding with BCL-2 family with the highest affinity. ABT-737 is an investigational anticancer agent which works through this mechanism. 70
ABT-737
A prototype BH3 only mimetic agent, ABT-737, is developed by Abbott Laboratories (North Chicago, IL, USA). 71 A clinical study showed that co-administrations of ABT-737 with other chemotherapeutic agents such as doxorubicin, gemcitabine, and cisplatin produced a synergetic effect in dedifferentiated thyroid carcinoma cells of various histological origins. 72 Moreover, on 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay and flow cytometry, ABT-737 along with radiation treatment induced synergistic cytotoxic effect with evidence of significant apoptosis. 73 In uterine cervical cancer cells, potentiation of apoptosis activator agent (Arsenic trioxide) was observed. The proposed mechanism is that arsenic trioxide reduces anti-apoptotic protein Mcl-1 in Caski cells and enhancement of ABT-737-induced apoptosis and caspase-7 activation. 74 These suggested that ABT-737 and related BH3 mimetic drugs, in combination or alone, may be of value as a new therapeutic option for dedifferentiated thyroid carcinomas. ABT-737 also appears to work through triggering platelet phosphatidylserine in a caspase-dependent manner. 72
BCL-2 inhibition by BH3 mimetic
These novel classes of anticancer agents work through inducing apoptosis by mimicking the activity of natural antagonists of BCL-2 and other related proteins. 70
Navitoclax
Navitoclax is an orally bioavailable, novel molecule that showed a promise starting from an early clinical phase. It targets and binds with greater affinity to multiple molecules including BCL-XL, BCL-2, BCL-w, and BCL-B, which are anti-apoptotic BCL-2 family proteins, and they are widely expressed in a variety of cancers, including prostate, colon, lung, and breast. A wide variety of cancers, including those of the lymph, breast, lung, prostate, and colon, are linked to tumor drug resistance. Inhibition of these apoptosis suppressors impedes their binding to the apoptotic effector’s Bak and Bax proteins, thereby promoting apoptotic processes in cells overexpressing Bcl-2, Bcl-XL, and Bcl-w. Similar to ABT-737, navitoclax has shown synergistic activity with taxanes in killing a variety of cancer cell lines. 75 In a preclinical study of SCLC xenograft models, administrations of 25–50 mg/kg navitoclax significantly attenuate tumor progression. 76
Venetoclax
The US Food and Drug Administration (FDA) approval of Venetoclax in 2016 for myeloid leukemia is a major milestone in cancer and apoptosis research. Venetoclax is also studied and approved for different types of cancer at different time points. 77 Venetoclax produces high overall treatment progression in high-risk subjects with chronic lymphocytic leukemia and complete response within significant proportion (20%) when it is given alone. It acts through binding to the BH3-binding groove of BCL-2 and displaces BH3-only proteins including Bim which are normally sequestered by the anti-apoptotic proteins. Thereafter, BH3-only proteins will be freed to activate the apoptosis effectors, Bax and Bak. ax/Bak activation that leads to mitochondrial outer membrane permeabilization through oligomerization. 78
CSC targeted therapy
CSCs are a small population that resist within the tumor despite appropriate chemotherapy. They control the dynamics of tumorigenesis by their ability of differentiation and rejuvenation. This characteristic greatly contributes to immune escape, metastasis, cellular heterogeneity, and tumor malignancy. The biological activities of CSCs are regulated by several pluripotent transcription factors, such as OCT4, Sox2, Nanog, KLF4, and MYC.19,79
An insightful understanding of CSCs redirects to a new way to build better therapeutic applications for cancer targeting this site. 19 For example, monoclonal antibodies (mAbs) such as rituximab that targets CSC-specific surface biomarkers have become an emerging technology for cancer therapy. Others, which target this site, include alemtuzumab, bivatuzumab, and talacotuzumab. Alemtuzumab is a recombinant humanized immunoglobulin monoclonal antibody directed against human CD52 (a glycoprotein expressed at high levels by both normal and malignant B and T lymphocytes). 80 The challenge with CSC targeted therapy is the heterogeneity and high plasticity of both CSCs and CSC tumor cells. Moreover, on normal stem cell populations, many surface markers are also widely expressed where they might play essential roles in tissue hemostasis and renewal. To date, attempts have been made to target stemness markers, such as STAT3, and as well as pathways that regulate malignant stemness, commonly JAK-STAT, Wnt/β-catenin, and hedgehog which promote stemness features in various cancer. 81
Mechanisms of anticancer drug resistance
Anticancer drug resistance is a big clinical challenge that yields many undesirable effects. Different possible mechanisms are contributed for anticancer drug resistance, for example, mutation or alteration of drug targets, an increase in drug efflux, drug inactivation and detoxification, interference with DNA replication, and impact on apoptosis are the main mechanisms faced by cancer therapy. 21
Expression of drug efflux pumps
The key causes for cancer therapy failure are multidrug resistance, which happened at the initiation or during the treatment when cells develop resistance to the treatment. The mechanism by which resistance developed is raised expression of medication efflux pumps, which is completed by P-glycoprotein. P-glycoprotein is encoded by the MDR1 gene and it is the ATP-binding transporters. P-glycoprotein plays a significant role in the maintenance and generation of colorectal cancer drug resistance. P-glycoprotein is a potential target to find novel compounds to counter multidrug resistance. MRP1 is an efflux pump that quickly squeezes out several anticancer medications from the targeted cancer cells in a different mechanism. Vandetanib and gefitinib can transport substrates for Breast Cancer Resistance Proteins and act as inhibitors of P-glycoprotein.7,21
Mutation or alteration of drug targets
Anticancer drug resistance can be affected by different drug targets at the molecular level. Some alterations of expression levels or the mutations of functional targets cause drug resistance. Recently, novel anticancer medications that targeted oncogenic signaling pathways have been synthesized. 81 A recombinant anti-HER2 agent (Trastuzumab) was the biological medication accepted and remained the gold standard for the management of HER2+. Regarding trastuzumab therapeutic efficacy, some HER2+ patients displayed acquired or intrinsic resistance to it. Protein overexpression and gene amplification are the possible resistance mechanisms to trastuzumab, usually as a result of HER2. 10
Impact on apoptosis
Drug resistance intermediated by anti-apoptosis is also the main MDR mechanism. The suppression of apoptosis is one common mechanism by which tumor cells developed drug resistance. B-cell lymphoma/leukemia-2 is known to be one of the anti-apoptotic proteins. 82 Proteins are critical in the cell’s life or death decisions and serve as rheostats for mitochondrial membrane stability. Moreover, B lymphoma cells undergo apoptosis following exposure to the topoisomerase II inhibitor etoposide. 21
Drug inactivation and detoxification
Tumor resistance can be developed during treatment, become apparent on re-treatment of the patient, or happen at the initiation of treatment. Glutathione S-transferase is needed for the development of drug resistance by direct detoxification. It reduces the concentration of anticancer medications through the glutathione (GSH)-conjugate export pump and distinct emphasis has been put on the function of Glutathione S-transferase in Phase II detoxification. 11
Cancer cells could develop cisplatin resistance by increasing the drug detoxification system via elevating the levels of intracellular scavengers like GSH in the treatment of cervical, ovarian, testicular cancer, and other types of cancer. Glutathione S-transferase is responsible for the catalyzation of the detoxification of cisplatin by its interaction with GSH. Other GSH-related enzymes could also have a significant role in this resistance process. 21
Interference with DNA replication
DNA damage of cancer cells is due to chemotherapeutic agents, and cancer could make changes in some genes to produce the mutator phenotype. Irinotecan enhances cancer cell death by interfering with the topoisomerase type1b enzyme (TOP1) on DNA, generating cytotoxic protein-linked DNA breaks.7,83
Topoisomerase-II is the principal target for several anti-carcinogens such as amsacrine, epipodophy, and anthracyclines. Through forming drug-Topo-II complexes in cancer cells, agents enable DNA strand breaks and affected DNA replication, sensitivity, and activity of Topoisomerase-II, which were vital in the development of drug resistance. DNA damage repair played a significant role in drug resistance, particularly resistance to poly(ADP-ribose) polymerase (PARP) inhibitors in the clinic. 21
Limitation
Even if this review has its strengths, such as the inclusion of many published articles and critically appraising the selected articles, it is not without limitations. The limitation of the present review is the complete reliance on previously published articles, and the use of terminology when searching evidence might be the other limitation.
Conclusion
Knowing about the pathophysiology of the disease, human gene sequence and the discovery of novel molecular targets is the core of modern medicine to conquer cancer therapy. In the new era of modern medicine, cancer therapy should switch bullet mass killing of cells to targeted selective of the malignant tumor cells and less/no damage to normal healthy cells. The development of target therapy requires the identification of cancer cell origination, growth, and survival with their respective mechanism of propagation to circumvent these malignant cells. New therapeutic agents with respective novel drug targets acting on genes, enzymes, and proteins are emerging and evolving new era of the clinical therapeutic center of cancer. Advanced anticancer agents such as angiogenesis inhibitors (bevacizumab and ramucirumab), DNA synthesis inhibitors (cytosine-β D-arabinofuranoside), transcription regulator (indarubicin), enzyme inhibitors (Z-endoxifen), and drugs acting on the targeted gene, enzyme, and proteins are promising drug research center to solve the current challenge of cancer therapy.
Footnotes
Acknowledgements
The authors would like to acknowledge University of Gondar and Addis Ababa University for providing free Internet access during reviewing of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.