
Abstract
Despite many advances in medical therapy for pulmonary arterial hypertension (PAH) over the past 20 years, long-term survival is still poor. Novel therapies which target the underlying pathology of PAH and which could be added to current vasodilatory therapies to halt disease progression and potentially reverse pulmonary vascular remodeling are highly sought after. Given the high attrition rates, substantial costs, and slow pace of new drug development, repositioning of “old” drugs is increasingly becoming an attractive path to identify novel treatment options, especially for a rare disease such as PAH.
We here summarize the limitations of current PAH therapy, the general concept of repurposing and repositioning, success stories of approved repositioned drugs in PAH as well as novel repositioned drugs that show promise in preclinical models of pulmonary hypertension (PH) and are currently tested in clinical trials. We furthermore discuss various data-driven as well as experimental approaches currently used to identify repurposed drug candidates and review challenges for the “repositioning community” with regards to funding and patent and regulatory considerations, and to illustrate opportunities for collaborative solutions for drug repositioning relevant to PAH.
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by progressive pulmonary vascular remodeling leading to increased right ventricular (RV) afterload, RV failure and, if untreated, early death.1,2
Many advances in medical therapies for patients with PAH have resulted in improved quality of life, exercise capacity, and survival; yet long-term survival remains poor. 3 While increased disease recognition of PAH has led to increased PAH prevalence, 4 high costs and cumbersome administration of current therapies complicate PAH treatment. Thus, in addition to the existing potent vasodilatory therapies, new therapies that target the underlying PAH pathology, modify the disease, halt progression, or induce regression of vascular remodeling are urgently needed.
The ideal medication for PAH would specifically target the pulmonary vasculature and the underlying disease mechanisms leading to PAH would reverse vascular remodeling and thereby reduce pulmonary vascular resistance (PVR), RV afterload, and strain on the failing RV. It would improve meaningful clinical outcomes, including survival, quality of life, and exercise capacity, while decreasing hospitalizations and Emergency Department visits. It would be easy for patients to administer, low in side effects, and low in cost. Unfortunately, current therapies for PAH do not meet these requirements of an ideal therapy, resulting in efforts to identify novel treatment targets and promising drug candidates.
Given the high cost and lengthy process of de novo drug discovery and the recent difficulty in developing new drugs for PAH, 5 repurposing existing drugs presents a desirable opportunity for PAH and other rare diseases. 6
The term “drug repurposing” was originally used for re-investigating drug-like molecules from pharma pipelines that had failed their initial indications, whereas the term “drug repositioning” was the application of already-approved drugs and compounds to treat a different disease. 7 At present, the terms “drug repositioning,” “drug repurposing,” “drug reprofiling,” “drug redirecting,” “drug rediscovery,” or “drug re-tasking” are all used interchangeably.6,7 Throughout this paper we will use the term “repositioned drugs” for the novel use of already-approved drugs.
Drug repurposing inherently involves an understanding of the pharmacology of the repurposed medication, as well as a detailed understanding of the disease pathophysiology. Drug repurposing can be a simpler and more cost-effective process than de novo drug development; therefore, it offers the opportunity for a timely and affordable avenue to identification of “new” therapeutic candidates. Epoprostenol, calcium channel blockers (CCBs), sildenafil, and tadalafil are successful examples of medications that have already been repositioned for the treatment of PAH. Currently, many ongoing investigator-initiated trials, as well as few pharmaceutically sponsored clinical trials, test repositioned drugs that target different aspects of PAH pathobiology: proliferation; mitochondrial dysfunction; hormones; altered metabolism; inflammation; and BMPR2 signaling. The purpose of this review is to highlight the advantages of drug repurposing and repositioning in PAH, to review promising drugs that are currently being tested in PAH, to discuss various data-driven and experimental approaches, to identify drug repurposing candidates, and to review challenges and opportunities for the “repositioning community” in PAH.
Approved therapies for pulmonary arterial hypertension: beneficial, yet not curative
Evaluating existing therapies for PAH in relation to the components of an ideal therapy.
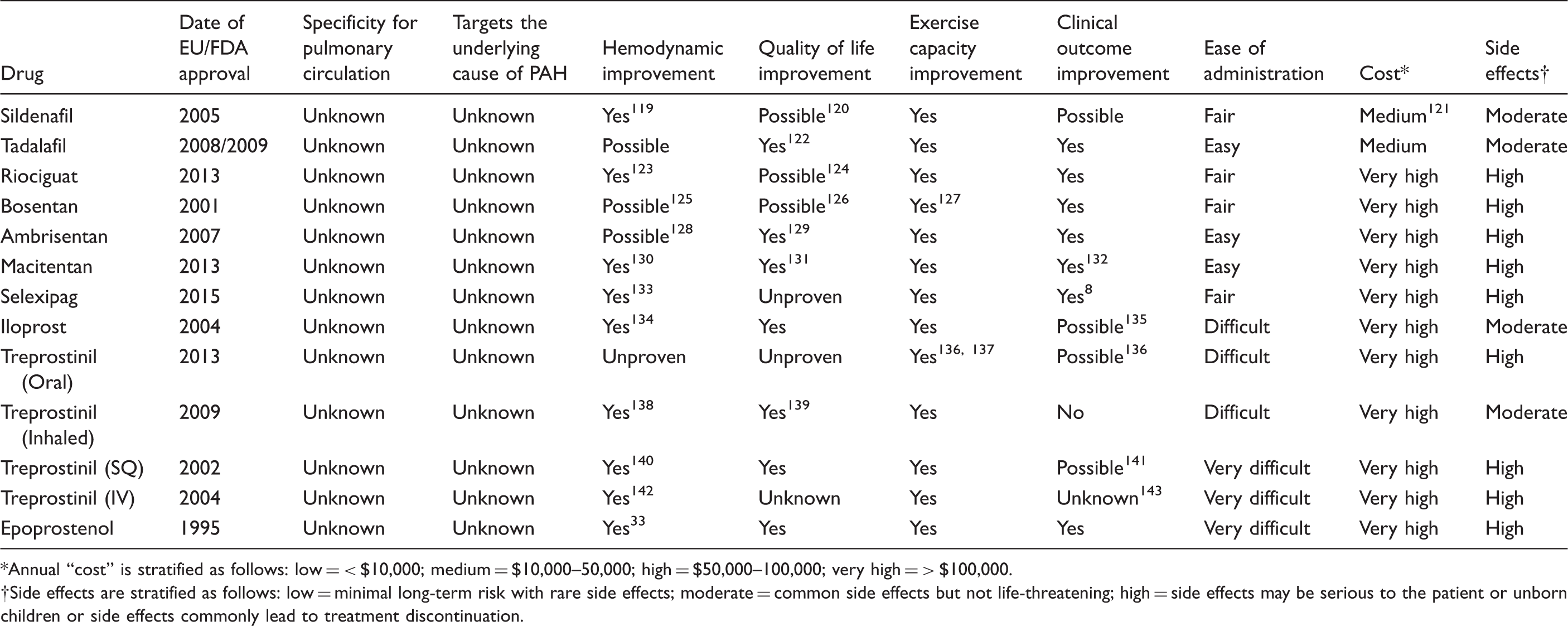
Annual “cost” is stratified as follows: low = < $10,000; medium = $10,000–50,000; high = $50,000–100,000; very high = > $100,000.
Side effects are stratified as follows: low = minimal long-term risk with rare side effects; moderate = common side effects but not life-threatening; high = side effects may be serious to the patient or unborn children or side effects commonly lead to treatment discontinuation.
Because the attributes of an ideal medication are not completely embodied in any current therapy, Table 1 is inherently incomplete and subjective. For example, when evaluating existing medications for their ability to “target the underlying cause of PAH,” we acknowledge that the pathophysiology of PAH is not fully understood. While endothelin-1 plasma levels are known to be elevated in patients with PAH, 10 and endothelin receptor antagonism (ERA) is known to be an effective treatment strategy, it remains unclear if the elevated endothelin levels are contributory to the underlying disease pathogenesis of PAH or whether they reflect a reaction to the underlying pathophysiology of the disease. 11 Similarly, modulation of the “prostacyclin pathway” through use of either prostacyclin analogues or prostacyclin IP receptor agonists has resulted in effective treatments for patients. However, while the prostacyclin pathway is dysregulated in those with PAH, 12 and prostacyclin has known antithrombotic and antiproliferative properties, 13 patients on prostacyclin do not have reversal of the underlying pulmonary arteriopathy that defines the disease pathogenesis.14,15 The lack of impact on the characteristic pathologic vascular lesion of PAH, combined with the current inability to implicate the prostacyclin pathway in the disease pathogenesis of PAH, has led to questions about whether prostanoid therapy truly effects the underlying pathogenesis leading to PAH.
Medications that manipulate the nitric oxide/cyclic guanylate monophosphate (NO/cGMP) pathway are also widely used in PAH management. As with the ERA and prostacyclin pathways, successful manipulation of the NO/cGMP pathway to improve patient outcomes has not been associated with a known effect on the underlying pathogenesis of PAH. While targeting NO/cGMP clearly results in pulmonary arterial vasodilatation, it remains unclear how this is linked to the underlying disease pathophysiology. Therefore, when taken as a whole, existing medications approved for PAH are clinically effective, but with uncertain effect on the underlying pathophysiology and with uncertain ability to impact the endothelial, smooth muscle, and adventitial proliferation that characterizes PAH.
In Table 1, we furthermore categorized current PAH targeted therapies according to the “severity” of their side effect profile. The purpose of including this category is to show that, in general, treatments of PAH have high side effect profiles that can be improved upon with future therapies. Riociguat and the ERAs (bosentan, ambrisentan, macitentan) all have significant teratogenicity which has led to the requirement for monthly pregnancy testing in women of childbearing age and formal risk evaluation mitigation strategy (REMS) programs. Because PAH affects women of childbearing potential disproportionately, this potential side effect has led to significant increases in cost, healthcare utilization, and patient inconvenience. Therefore, these medications were labeled “high risk” based on their teratogenic risk alone, irrespectively of additional other side effects such as liver toxicity, fluid retention, and anemia. Several medications in the prostanoid group were also labeled “high risk” based on the high percentage of significant side effects that patients experience that commonly limits up-titration, such as severe headaches, nausea, vomiting, and foot pain; 16 there is also the high percentage of patients on subcutaneous treprostinil who stop therapy due to site pain 17 and the serious nature of rare but life-threatening bacteremia associated with intravenous medications. 18
The categories from Table 1 labeled “cost” and “ease of administration” also deserve a brief explanation. Unfortunately, the cost of existing therapies for many PAH therapies remains very high, often > $100,000 per year. Because of the high costs of current PAH therapy, patients suffer from high co-payments and insurance companies require prior authorization before initiation of therapy (which often leads to treatment delay or inability to provide treatment). The interaction between provider's office, insurance companies, co-payment assistance foundations, pharmacies, and the patient is a frequent source of frustration for patients and a significant resource utilization for the provider's office. The category “ease of administration” was developed to show the current difficulty of taking PAH therapy. Any medication that is oral and requires “once daily” administration was labeled “easy,” while oral medications requiring “twice a day” or “three times a day” administration were labeled “fair.” Oral treprostinil, despite its “three times a day” administration, was labeled as “difficult” due to its additional requirements of being taken every 8 h with administration of food, as well as the complexity of its titration schedule. The remaining “difficult” medications have intricate delivery systems requiring frequent assessment, possibly excluding those patients with fewer resources or lower health literacy.
In summary, current therapies have been shown to benefit patients with PAH with regards to exercise capacity, quality of life, and general morbidity from PAH. However, they generally do so with a high cost to the healthcare system and a high side effect profile. In addition, it is unclear if existing therapies target specifically the pulmonary circulation and the underlying pathways that contribute to the pathogenesis of PAH. Lastly, we note that medications approved within the past 12 years share a common mechanism of action (cGMP activation, ERA, prostanoids) yet predominantly only vary in their modes of application—inhalation, oral and subcutaneous delivery—and are therefore labeled “me-too” drugs. 19 No new PAH medication has been approved in several years, supporting the significant need for novel therapies for PAH.
Prior examples of drug repurposing in pulmonary arterial hypertension
There are different pathways through which new therapies for PAH can make their way into clinical practice. New drug development, or de novo drug development, involves taking a product from its molecular origination through basic science development, preclinical and clinical development, and regulatory approval. As such, it is not uncommon for the process of de novo drug development to take 12–15 years and cost from the low $100 million to in excess of $1 billion. 20
The process of drug repositioning, which often involves the use of generic therapies already approved by the FDA, will typically remove the steps of molecular origination, basic science development, in vitro and in vivo screening, chemical optimization, toxicology, bulk manufacturing, formulation development, and possibly phase I clinical trials. Removing these steps improves the chance of a new drug application gaining market approval. Only 10% of new drug applications gain approval, while 30% of repositioned drugs gain approval. 21 Therefore, a repositioned medication can often make its way to the market after 4–7 years and with significantly less cost, estimated at $20–100 million dollars on average, 6 a fraction of the cost of average de novo drug development. As Chong et al. point out, because existing drugs have known pharmacokinetics and safety profiles and are often approved by regulatory agencies for human use, any newly identified use can be rapidly evaluated in phase II clinical trials, which typically last two years and cost $17 million.22,23
Drug repurposing can have a goal of either philanthropic use or use for commercialization. This means that, instead of aiming for commercialization of a new indication, philanthropic repurposing seeks to provide enough scientific evidence to allow clinicians to decide if a new therapy can be used to treat their patients in “off label” use. As such, philanthropic repurposing of medications offers the least time and expense, but it can suffer from lower usage by physicians and difficulties in reimbursement from payers.
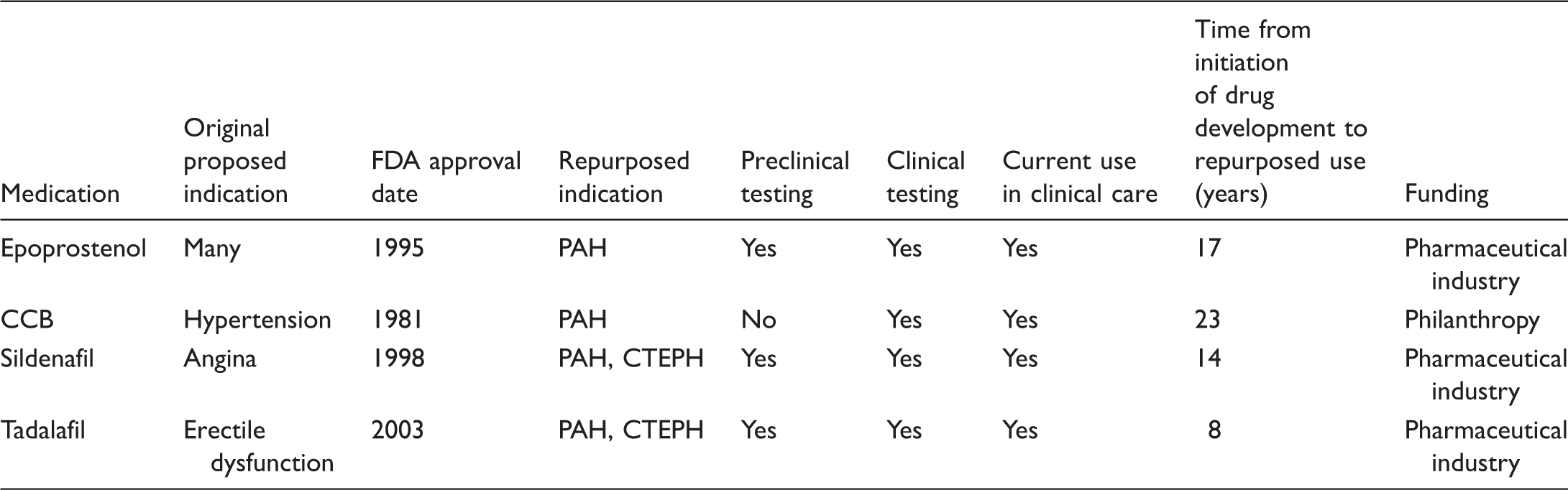
To highlight some of the difficulties and nuances that have made drug repurposing historically difficult in PAH, we will review the process of repurposing through examples from existing PAH therapies (Table 2).
The path to FDA approval of epoprostenol for PAH was long and arduous. For decades stretching from the 1950s, researchers tried to delineate the pathways by which prostaglandins are formed and discovered that prostaglandins resulted from the breakdown of arachidonic acid. Prostacyclin was discovered in 1976, 24 for which John Vane was later rewarded the Nobel Prize. Prostacyclin was noted to inhibit platelets and dilate arterial beds. 25 This exciting discovery led to a rush to make a synthetic prostacyclin and to find uses for prostacyclin in medicine. Prostacyclin was proposed as a therapy for multiple diseases, such as systemic hypertension 26 and coronary artery disease, 27 as well as an anticoagulant to assist with dialysis. 28 Development of a medication for use in patients was difficult, due to the short half-life and instability of synthetic prostacyclin under many conditions. 29 Eventually, further preclinical testing showed that prostacyclin infusion led to a reduction in PVR, 30 thus leading to the potential to repurpose epoprostenol (synthetic prostacyclin) as a treatment for PAH. In 1984, a landmark case report from Higenbottam et al. showed that continuous intravenous infusion of epoprostenol in the outpatient setting led to clinical and hemodynamic improvement in a PAH patient. 31 This led to a phase II, randomized study of 24 idiopathic PAH (IPAH) patients, where a significant hemodynamic improvement was found in those patients treated with epoprostenol. 32 This result was followed by a phase III study which showed improved mortality and exercise capacity in those patients treated with epoprostenol, leading to the first FDA approval for a drug for the indication of PAH in 1995. 33 The cost of epoprostenol therapy for PAH was and is very high due to multiple reasons: PAH is a rare disease, the product development was associated with a considerable expense and the repurposing efforts of epoprostenol for PAH was largely driven by commercial interests of the pharmaceutical industry. Since the mid-1990s, despite being a generic product for many years, there has not been significant competition in manufacturing to influence costs. Improvements in the molecular stability of epoprostenol at room temperature have led to new formulations, but the pricing of these generic products has remained very high (as improvement of a product at an equivalent cost will drive consumers toward the new product). The repurposing of epoprostenol has taught us that the intent to commercialize via the pharmaceutical industry typically leads to products with very high cost. As discussed above, the high cost of PAH medications has led to a need for more affordable alternatives.
The story of repurposing CCBs for PAH serves as a contrasting example to epoprostenol (philanthropic motive versus commercialization motive). Initially developed as antihypertensive therapies in the 1960s, CCBs decrease intracellular calcium as a mechanism to promote smooth muscle vasodilatation. 34 By the 1980s, PAH was recognized as a distinct clinical entity in need of therapy and CCBs were identified as a potential treatment based on their success as antihypertensives which vasodilate the systemic circulation. Clinical studies in patients with PAH progressed to a pivotal publication showing improved survival in a cohort of patients with PAH using high-dose CCBs. 35 As the purpose of this research was philanthropic, it did not lead to a new FDA indication for PAH. As CCBs were low in cost at the time of repurposing, they have remained a cost-effective therapy for PAH. The low cost yields a competitive disadvantage for CCBs as they do not have nearly the marketing and product support of more expensive pulmonary vasodilator therapy. However, in the current treatment of PAH, CCBs remain effective treatments for a small cohort of patients with PAH who have a sustained response to these medications after a favorable vasodilator challenge during the diagnostic right heart catheterization (RHC). 36
A third example of existing drug repositioning in PAH are the phosphodiesterase 5 inhibitors (PDE5i). In the 1980s, it was well recognized that cGMP activation resulted in decreased intracellular calcium, causing vascular smooth muscle relaxation. Phosphodiesterase 5 (PDE5) was identified as a catalyst for cGMP breakdown and therefore a potential target for smooth muscle vasodilatation. 37 Sildenafil emerged as a promising potential therapy; by 1991, early studies were underway to evaluate its efficacy in treating angina. 38 Interactions with nitrates and other issues led to these efforts being abandoned, but retrospective clinical analysis revealed a common side-effect of erectile dysfunction (ED) which led to its repositioning (with FDA approval in 1997) as the leading product in the ED market with global sales of > $2 billion in 2012. 6 Soon after its serendipitous repurposing for ED, physiologic evidence emerged linking sildenafil and PDE5 inhibition to the pulmonary circulation.39,40 Ultimately, the SUPER-1 study (referenced in Table 1), at its time the largest clinical trial in PAH, was initiated in 2002 and its favorable outcome led to approval in 2005. While sildenafil was repurposed with commercial intent, it went “off patent” in 2012. Due to its easily reproducible pill form, several generic competitors have developed sildenafil; this competition has slowly decreased costs and led to sildenafil as the only FDA-approved PAH therapy with a “moderate cost” label in Table 1.
New tools to expand and re-design repositioning and repurposing in pulmonary arterial hypertension
In 2004, Ashburn and Thor wrote their landmark article “Drug repositioning: identifying and developing new uses for existing drugs,”
21
in which they outlined the growing productivity gap in the biopharmaceutical industry and the need for novel drug development strategies. The authors pointed out that over the past 20 years, the output of the biopharmaceutical industry has not kept pace with the enormous increases in pharmaceutical research and development (R&D) spending despite the fact that pharmaceutical companies invested prodigious amounts in novel discovery technologies, such as structure-based drug design, combinatorial chemistry, high-throughput screening (HTS), and genomics. Attempts to reduce pharmaceutical R&D timelines were often associated with increasing risk and later failure of the drug. As we demonstrated above, drug repurposing in PAH has been largely opportunistic and serendipitous; once a drug was found to have an off-target effect or a newly recognized on-target effect, it was taken forward for commercial exploitation.
6
Indeed, the most successful examples of drug repurposing so far have not involved a systematic approach. More recently, a combination of systematic computational as well as experimental approaches have been used to best identify the right drug for an indication of interest with a high level of confidence (Fig. 1). Examples of computational approaches are “signature matching” of a drug against another drug, disease, or phenotype, whereby the signature could be derived from transcriptomic (RNA), proteomic, metabolomic data, chemicals structures, or adverse event (AE) profiles.6,41,42 The goal would be to identify drugs that could reverse the expression pattern of a given gene set that characterizes a particular disease, which is called “signature reversion principle.” Drug–drug similarity approaches try to identify drugs from different classes that might have a shared mechanism, called the “guilt by association” approach, which then could help identify alternative targets of existing drugs and uncover potential off-target effects that could be further investigated for therapeutic use.
41
Another promising approach is combining our knowledge of genome-wide association studies (GWAS) with drug target-databases such as PharmGKB and Drugbank, the rationale being that genes that were associated with a disease trait were more likely to code for proteins that were “drugable” or “biopharmable” than the rest of the genome.
43
Furthermore, retrospective clinic analysis using electronic health records might uncover many “off-target” effects of drugs by looking at the side effect profile.
44
Evaluation of combinations of healthcare records and genomic analysis have led to all sorts of repurposing research leads.
45
Examples for experimental systematic approaches to identify promising drug candidates are binding assays (to identify relevant target interactions) as well as phenotypic high-throughput compound screening using in vitro or in vivo disease models.46,47
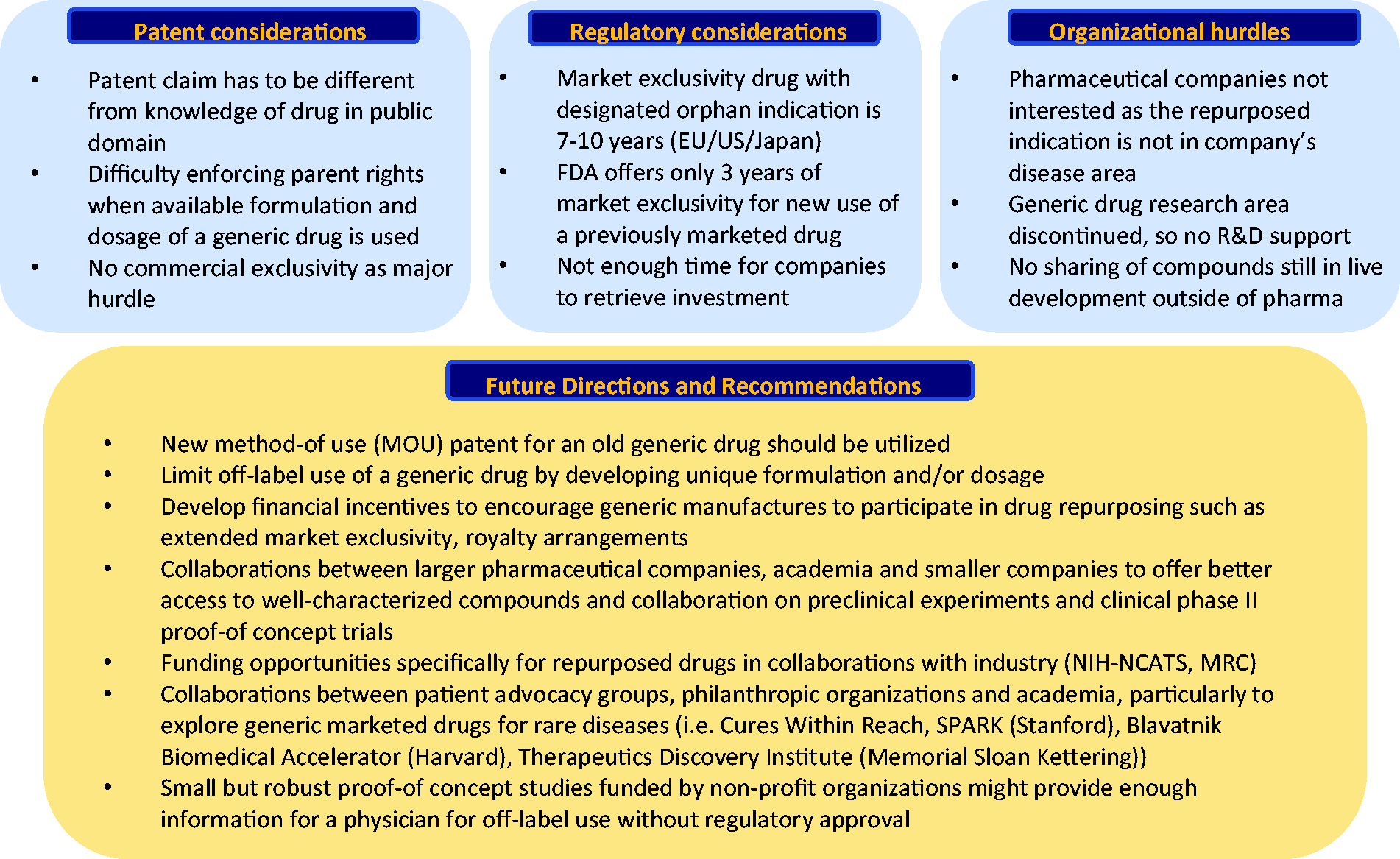
Current and future approaches for drug repositioning: serendipity vs. systemic approaches. While traditionally drug repurposing mostly relied on serendipity, novel computational and experimental HTS approaches and combinations of both offer promising avenues to identify novel drugs for repurposing strategies. HTS, high-throughput screening; HER, electronic health record. Barriers to drug repurposing and future directions This figure lists the current barriers to a successful drug repurposing and repositioning including patent, regulatory and organizational hurdles and summarizes future directions and recommendations (modified from Pushpakom et al. Nature Reviews 2018). EU, European Union; US, United States; R&D, Research and Development; NIH-NCATS, National Institute of Health – National Center for Advancing translational Sciences; MRC, Medical Research Council; SPARK, Translational Research Program of Stanford.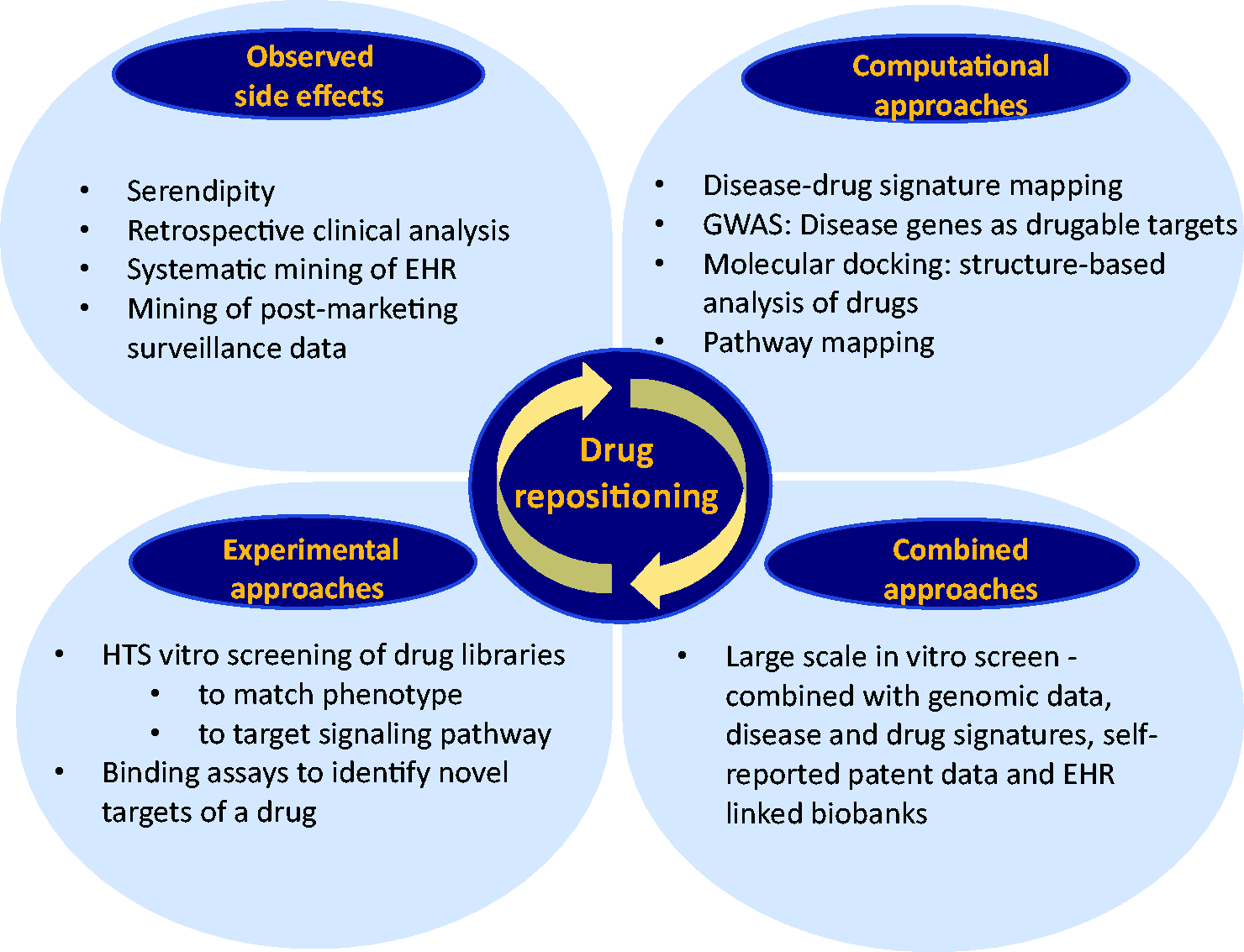
At present, there are > 2000 medications approved for generic use and there are > 3000 nutriceuticals in use. These therapies have thousands of different mechanisms of action that, when carefully selected to match the existing knowledge about an existing disease, could lead to a promising therapy. In an era of PAH research in which we are gathering genomic, proteomic, and metabolomic information that could help us to personalize therapies for our patients, the multiple mechanisms of action available through repurposing existing therapies offers tremendous promise. Importantly, as existing PAH therapies are very expensive, repurposing medications are often generic and less expensive to develop, and, as such, could offer much more affordable treatment options and perhaps wider safety margins and lower side effect profiles.
Drug repurposing and repositioning in pulmonary arterial hypertension: promising novel approaches
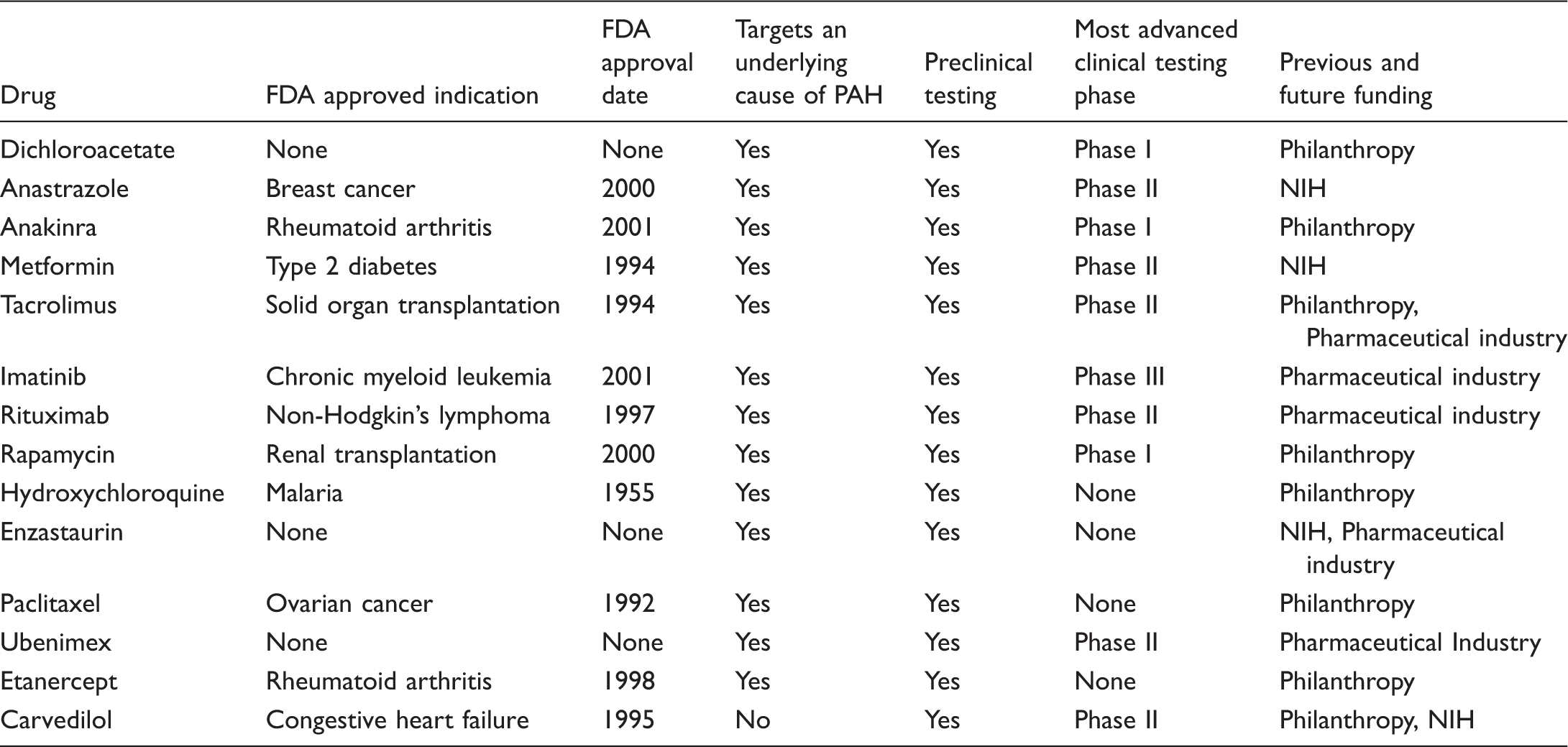
Approved PAH therapies that were repurposed or repositioned for use in PAH.
Characteristics of novel drug candidates being repurposed and repositioned for clinical use in PAH.
Targeting proliferation
Imatinib
Imatinib was the first drug without vasodilatory properties that was tested for its potential to reversal vascular remodeling in PH. Imatinib is a tyrosine kinase inhibitor that specifically blocks abl, c-kit, and the platelet-derived growth factor (PDGF) receptor and is known to block the brc-abl activity in chronic myelogenous leukemia (CML) for which imatinib is FDA-approved.
Given that PDGF signaling is associated with smooth muscle cell proliferation and is highly increased in PH, imatinib was “repurposed” to treat experimental PH and impressively reversed advanced pulmonary vascular disease in two animal models of PH, the monocrotaline rat model and the hypoxia mouse model, 48 by inhibiting pulmonary artery smooth muscle cell (PASMC) proliferation. Subsequently, imatinib (200 mg daily) was administered to an end-stage PAH patient awaiting lung transplantation who showed an impressive improvement after three months of treatment, as indicated by improved exercise capacity, improved hemodynamics and PVR, and an improved functional class (FC; New York Heart Association [NYHA] class II), an effect that was sustained after six months of treatment. No side effects were apparent. 49 Building on this, a phase II, 24-week, randomized, double-blind, placebo-controlled proof-of-concept pilot study enrolled 59 patients (imatinib [n = 28]; placebo [n = 31]) in FCs II–IV. This study found a significant decrease in PVR (imatinib –300 ± 347 versus placebo –78 ± 269 dynes·s·cm−5, P < 0.01) and an increase in cardiac output. Post hoc subgroup analyses suggested that patients with greater hemodynamic impairment may respond better than patients with less impairment. Enthusiasm was tapered, however, due to fairly poor tolerance with a high incidence of serious adverse effects (39% in imatinib versus 21% in placebo). 50
In the following IMPRES trial (Imatinib in PAH, a Randomized, Efficacy Study), patients with PAH were randomized in a 1:1 ratio to imatinib or placebo once daily. Imatinib improved exercise capacity, as measured by 6-min walk distance (6MWD) and hemodynamics in patients with advanced PAH, with a significant increase in cardiac output, but had little effect on mean pulmonary artery pressure (mPAP). 51 Thus, although PVR decreased, it was not clear whether there was regression of the pulmonary vasculopathy, despite imatinib being an antiproliferative agent. Serious adverse events (SAEs), most particularly subdural hematoma, were common, and in general the drug was poorly tolerated, with frequent discontinuation of imatinib. Early drop-outs, missing dose adjustment for non-cancer patients, inadequate counseling of expected side effects of patients and study personnel, and missing upfront characterization of potential responders were all factors that were discussed as having contributed to the failure of the clinical trial.
It was concluded that a better understanding of the pathways involved in the efficacy and safety aspects of imatinib would be paramount for the design of more targeted and better tolerated agents as a personalized PAH medicine approach. Until then, it was suggested that healthcare professionals refrain from offering compassionate imatinib therapy to PAH patients. 52
Targeting mitochondrial dysfunction
Dichloroacetate
PAH involves progressive obliteration of the pulmonary arterioles and is characterized by cellular proliferation and impaired apoptosis. 53 These proliferating cells have suppressed mitochondrial glucose oxidation with a resultant upregulation of glycolysis to provide cellular energy. 54 Mitochondrial suppression, in turn, is associated with impaired apoptosis and accelerated cellular proliferation. 55 Pyruvate dehydrogenase kinase (PDK) suppresses mitochondrial activity and is known to be increased in patients with PAH. 56 These discoveries indicate that PDK may be a novel therapeutic target for those with PAH, as blocking its activity at the level of the mitochondria could halt the cellular proliferation that characterizes the disease. Cellular mitochondrial activity is also dependent on normal functioning of the enzymes sirtuin 3 (SIRT3) 57 and uncoupling protein 2 (UCP2), 58 and deficiencies of these enzymes are both common in the general population and associated with development of PAH.59,60 Therefore, the effectiveness of PDK inhibition could be limited to patients with normal levels of SIRT3 and UCP2.
Dichloroacetate (DCA) is a PDK inhibitor that has been used to treat patients with the mitochondrial disease pyruvate dehydrogenase (PDH) deficiency. 61 The anti-proliferative effects of DCA have also led to its early phase testing in advanced solid tumors. 62 Based on the plausibility that DCA could impact the underlying cellular proliferation of PAH through PDK inhibition and the known safety profile of DCA through its use in other disease states, a clinical study was performed in PAH patients using DCA. In this open label study, 20 patients with PAH who were stable on PAH-approved therapies were given varied doses of DCA. At the highest dose of DCA administered, four patients developed peripheral neuropathy and withdrew from the study, so the final analysis involved 16 patients. DCA was administered over four months and patients had significant improvement in mPAP (P < 0.05) and PVR (P < 0.05) on RHC, as well as 6MWD (P < 0.05). While these results in themselves are clinically meaningful in patients on background therapy, the investigators noted significant inter-individual variability. Due to the potential impact of SIRT3 and UCP2 variants on DCA functioning, the level of SIRT3/UCP2 functioning was assessed in all patients and scored from 0 (low level of functionality) to 3 (high level of functionality). Lower functionality scores were associated with significantly stronger responses to DCA compared to those with higher functionality scores. These results suggest that SIRT3/UCP2 functionality confers genetically driven resistance to DCA. In the next step of a placebo-controlled study, this SIRT3/UCP2 functionality score could serve as a biomarker for use in patient selection, and in this way serve as a model for the use of precision medicine in PAH.
Targeting hormones and altered metabolism
Anastrazole
The long-standing estrogen paradox of PAH is that female gender is a very strong risk factor for the development of PAH, but female PAH patients have improved prognosis compared to men.4,63 The development of PAH is associated with estrogen metabolites, and aromatase (which accounts for most of the estrogen production in men and postmenopausal women) activity is associated with both circulating estrogen metabolites and risk for developing PAH. 64 Aromatase is also significantly increased in PASMCs of PAH patients. 65
Anastrazole (AN) is an aromatase inhibitor and is approved for treatment of breast cancer. It has been widely used in this indication for 20 years and it is generally well tolerated. 66 In an animal model of PAH, AN was found to improve pulmonary hemodynamics as well as RV function. 67 Based on the above physiologic rationale and preclinical data, a placebo-controlled, multicenter (two centers) study of AN in PAH was performed with a three-month treatment duration. 68 While the primary outcome of the study (change in tricuspid annular plane systolic excursion, or TAPSE, from echocardiogram) was not significant, the results of secondary outcomes from this exploratory study were intriguing. Not only were estradiol levels significantly decreased (P < 0.003) in the AN treated group as expected, but 6MWD was significantly improved (P = 0.02) by 26 m in the AN group. In addition, AN was well tolerated with no SAEs. Based on this pilot study, a multi-center, phase II study (PHANTOM) is currently underway (www.clinicaltrials.gov NCT03229499). In PHANTOM, the primary outcome is 6MWD over six months; secondary outcomes include comparisons of hemodynamics, RV imaging, biomarkers, and quality of life. If outcomes from this phase II study are significant, AN could offer an affordable, well-tolerated therapy that specifically targets the disease pathogenesis.
Metformin
Hyperglycemia and insulin resistance are associated with worse survival among patients with PAH. 69 Both hyperglycemia and insulin resistance have been associated with many biochemical mediators that are implicated in the pathogenesis of PAH. This includes impairment of nitric oxide synthase, 70 BMPR2 deficiency, 71 reduced levels of peroxisome proliferator-activated receptor gamma (PPARγ), 72 and increased endothelin levels. 73
Metformin is a hypoglycemic medication which also inhibits aromatase (see above discussion on AN) and ameliorates PAH in an animal model. 74 In addition, metformin ameliorates RV lipid deposition in an animal model of PAH. 75 Based on the escalating knowledge implicating these pathways in PAH, and the potential ability of metformin to favorably impact these metabolic pathways, a phase II clinical study is underway to assess the impact of metformin in patients with PAH (www.clinicaltrials.gov NCT01884051).
Targeting inflammation and immunity
Rituximab
Rituximab is a chimeric mouse anti-human IgG antibody that binds to CD20, an integral transmembrane protein expressed on the surface of normal and malignant B-lineage cells. By binding to CD20, rituximab leads to apoptosis of B-lymphocytes with antibody- and complement-dependent cytotoxicity. This mechanism of action leads, in most patients, to a selective peripheral B cell depletion for >24 weeks. 76 Rituximab was the first clinically successful antibody in oncology and was approved by the FDA in 1997 for the treatment of B cell lymphomas, particularly Non-Hodgkin's lymphoma. 77 It was later approved for rheumatoid arthritis and anti-neutrophil cytoplasmic antibodies-associated vasculitis and is nowadays also used off-label for other rheumatological diseases in patients with systemic sclerosis, Sjögren's syndrome, and systemic lupus erythematosus. 76
Regulatory T cell (Tregs) deficiency, dysregulated B cells, and pathogenic endothelial autoantibodies have been implicated in the pathogenesis of PAH. Athymic nude rats (which lack Tregs) exposed to Sugen/hypoxia showed pulmonary B cell accumulations and anti-endothelial cell antibody deposition in the pulmonary vasculature, resulting in severe experimental PH. Immune reconstitution with healthy Tregs prevented B cell accumulation and anti-endothelial cell antibodies and the development of PH. 78 While investigators are pursuing therapeutics that might increase Tregs, reducing B-cells with rituximab has anecdotally been reported to be effective in connective tissue disease-related PAH, including scleroderma-associated PAH. The ASC01 study (www.clinicaltrials.gov NCT01086540) is a NIH-funded, double-blind, placebo-controlled phase II randomized controlled trial (RCT) to evaluate the safety and efficacy of rituximab (two doses given two weeks apart) on disease progression in individuals with systemic sclerosis-associated PAH. The primary endpoint is the change in PVR at 24 weeks; secondary endpoints include RV function measured by magnetic resonance imaging (MRI). 79 While the recruitment is finished, the results have not yet been published.
Anakinra
Inflammation is well accepted as part of the pathogenesis of PAH and many associated causes of PAH (systemic sclerosis, HIV, schistosomiasis) are highly inflammatory. Interleukin-1 (IL-1) and interleukin-6 (IL-6) are inflammatory cytokines and their levels correlate with survival in PAH. 80 In addition, in the setting of BMPR2 dysfunction, IL-1 can act as a pulmonary-specific, disease-modifying stimulus and is thereby directly linked with PAH pathogenesis. 81 In an animal model of PAH, an antagonist against the IL-1 receptor was associated with a reduction in PA pressure, as well as improved RV function. 82
Anakinra is a recombinant form of the naturally occurring IL-1 receptor antagonist that has been approved for use in rheumatoid arthritis and autoinflammatory conditions. 83 In patients with reduced left ventricular ejection fraction (LVEF) and elevated levels of systemic inflammation (serum C-reactive protein [CRP] level >2 mg/L), treatment with anakinra has been tested in early phase studies in patients suffering from an admission for acutely decompensated heart failure 84 as well as in patients who have been recently admitted for the same, 85 effectively reducing inflammatory biomarkers in both populations with promising improvements in exercise capacity in the latter group (REDHART). Similarly, in a sub-study of the large CANTOS trial, patients who met CANTOS criteria but who also had reduced LVEF had an improvement in exercise capacity and ejection fraction after treatment with canakinumab. 86
Based on the above physiologic rationale and preclinical data, an open-label, proof of concept study was recently performed in PAH patients. 87 Six patients were given anakinra as a daily injection over two weeks. Anakinra was well tolerated without any SAEs and it significantly reduced serum CRP (P = 0.05). Reduction in CRP was associated with improvement in exercise capacity as measured by maximal oxygen consumption (P = 0.04). There was also significant improvement in quality of life as measured by the Minnesota Living with Heart Failure Questionnaire (P = 0.05). The next step of investigation for anakinra in PAH would likely involve multiple centers in a placebo-controlled trial that would utilize biomarkers (CRP) to assist with patient selection. While a recent investigation of IL-6 antagonism in PAH (TRANSFORM-UK) 88 is not likely to progress to further investigation, we note that this should not dampen enthusiasm for more robust IL-1 antagonism.
Rapamycin
Rapamycin was discovered as an antifungal agent in 1975 on the Easter Island Rapa Nui, hence the name, 89 and was subsequently found to have immunosuppressive properties leading to its approval as an immunosuppressant after solid organ transplantation 90 as well as an antiproliferative agent applied to coronary stents to reduce local restenosis. 91 Besides its immunosuppressive activity, rapamycin and its analogs have additional therapeutic potentials, including antitumor, neuroprotective/neuroregenerative, and lifespan extension activities. Rapamycin inhibits mammalian target of rapamycin (mTOR) by associating with its intracellular receptor FK506-binding protein 12 (FKBP12). 92 mTOR is a major regulator of cellular metabolism, proliferation, and survival that is implicated in various proliferative and metabolic diseases, including obesity, type 2 diabetes, hamartoma syndromes, and cancer. 93 Emerging evidence suggests a potentially critical role of mTOR signaling in pulmonary vascular remodeling. Rapamycin treatment, while having strong antiproliferative properties and preventing the development of pulmonary vascular remodeling, is not sufficient to induce apoptosis in pulmonary artery vascular smooth muscle cells. 93 A phase 1, 16-week clinical trial of inhaled albumin-bound rapamycin for patients with severe PAH is currently underway, with the primary endpoint being “number of participants with treatment-related adverse events” (www.clinicaltrials.gov NCT02587325).
Ubenimex
Leukotriene B4 (LTB4) has long been recognized as a chemoattractant for many inflammatory cell types observed in PAH, such as T and B lymphocytes, macrophages, and neutrophils. 94 In both clinical tissue and the Sugen/athymic rat model of severe PH, accumulated macrophages expressed high levels of leukotriene A4 hydrolase (LTA4H), the biosynthetic enzyme for LTB4. 95 Macrophage-derived LTB4 directly induced apoptosis in pulmonary artery endothelial cells (PAECs). Furthermore, LTB4 induced proliferation and hypertrophy of human PASMCs. It was therefore hypothesized that macrophage-derived LTB4 might play a role in PH pathogenesis and inhibiting LTB4 might be a therapeutic target in PAH. Bestatin, a LTA4H inhibitor that blocks LTB4 formation, 96 reduced serum LTB4 levels, prevented PAEC injury, and improved established PH in the Sugen/athymic rat model. As an immunomodulatory agent, Bestatin (Ubenimex) was used in the 1990s as treatment for hematological cancers and was subsequently tested in solid tumors. 97 Initial clinical data come from the “Study of Ubenimex in Patients with Pulmonary Arterial Hypertension (WHO Group 1) (LIBERTY),” a proof-of-concept, phase 2, multicenter, randomized, double-blind, placebo-controlled study. This study compares ubenimex with placebo in patients with PAH (World Health Organization [WHO] Group 1) and have a WHO/NYHA FC of II or III and was completed in 2018. The primary objectives for the study were: (1) to evaluate the efficacy of ubenimex in patients with PAH; and (2) to evaluate the safety and tolerability of ubenimex in patients with PAH. While the results of the study are not published yet, a press release this year suggested that the primary endpoint, a reduction in PVR, was not achieved, yet that subgroup analyses and characterization of responders are pending.
Targeting the bone morphogenetic protein receptor 2 (BMPR2) pathway
Tacrolimus
More than 70% of patients with familial PAH (FPAH) and 20% of patients with IPAH have heterozygous mutations in the gene which codes for the bone morphogenetic protein receptor 2 (BMPR2). 98 Furthermore, less frequently observed gene mutations in FPAH (ENG, ALK1, SMAD9, BMP9) also belong to the BMP pathway. 98 Even in non-familial PAH, dysfunction in BMPR2 or downregulation of the receptor is present, 99 suggesting BMPR2 signaling and expression to be a promising treatment target for multiple forms of PAH. 100 To identify BMPR2 activators that could be readily tested in the clinic, we performed a high-throughput screen (HTS) of FDA-approved drugs and bioactive compounds using the expression of the inhibitor of differentiation (ID1), a downstream target of BMPR2, as a readout. 47 Applying this systematic approach to identify drugs that could be repurposed to treat PAH, we identified the immunosuppressive drug FK506 (Tacrolimus) as the best BMPR2 signaling activator. Low-dose FK506 improved endothelial dysfunction of vascular cells from PAH patients, prevented hypoxia-induced experimental PH, and reversed experimental PH in the monocrotaline and sugen/hypoxia/normoxia rat model by increasing BMPR2 signaling through a dual mode of action: removing the TGF-β pathway inhibitor FKBP12 as well as inhibiting calcineurin. Given that tacrolimus has been used as an immunosuppressive therapy after solid organ transplantation for > 30 years, the pharmacokinetics, side effect profile, and dosing were already well-studied and established. It was decided to use tacrolimus clinically at a low dose (1–5 ng/mL trough blood level) given that these doses already increased BMPR2 signaling in vitro and, furthermore, out of fear to induced immunosuppression in patients prone to blood stream infection due to indwelling catheters for prostacyclin therapy. Tacrolimus was first employed for compassionate use in three advanced PAH patients on maximal medical therapy who were listed for lung transplantation. 101 All three patients stabilized after one year of low-dose tacrolimus treatment (trough level 1.5–2.5 ng/mL) in terms of symptoms, 6MWD, NT-proBNP, and RV function by cardiac MRI. A 16-week, placebo-controlled RCT included 23 stable PAH patients and showed that tacrolimus (blood level 1–5 ng/mL) was well tolerated. 102 While the study was not intended to assess efficacy but rather proof-of-concept, some patients (targeting blood levels of 3–5 ng/mL) had a significant increase in BMPR2 expression in peripheral blood mononuclear cells, supporting future efficacy studies in a larger patient cohort. Due to the ease of clinical translation of a repurposed drug, treatment with tacrolimus is the only strategy to increase BMPR2 signaling – as of now – that has been clinically tested in PAH patients, despite multiple promising compounds in pharmaceutical development. Given that tacrolimus is an immunosuppressive drug, it cannot be excluded that part of the beneficial effect observed in the preclinical studies and compassionate use patients is in part due to an effect on the immune system.
Paclitaxel and hydroxychloroquine
Two FDA-approved drugs – the chemotherapeutic drug paclitaxel and the immunosuppressive and anti-malaria medication hydroxychloroquine – have been shown in preclinical studies to improve experimental PH, both by modulating – among other targets – the BMPR2 pathway.103,104 Paclitaxel increases the transcription factor Forkhead box O 1 (FoxO1), a key regulator of cellular proliferation. Pharmacological inhibition and genetic ablation of FoxO1 in smooth muscle cells reproduced PH features in vitro and in vivo. Either pharmacological reconstitution of FoxO1 activity using intravenous or inhaled paclitaxel, or reconstitution of the transcriptional activity of FoxO1 by gene therapy, restored the physiologically quiescent phenotype of PASMCs in vitro, linked to changes in cell cycle control and BMPR2 signaling, and reversed vascular remodeling and right-heart hypertrophy in vivo. 103 Hydroxychloroquine, as well as chloroquine, prevented the development of experimental PH, RV hypertrophy, and vascular remodeling in rats exposed to monocrotaline, and prevented progression of established PH in this model by influencing autophagy pathways and inhibiting of lysosomal degradation of BMPR2. 104 Neither medication has been tested clinically in PAH patients. Given that hydroxychloroquine is a frequently prescribed drug for autoimmune diseases such as lupus, rheumatoid arthritis, psoriasis, and occasionally scleroderma, it would be an ideal drug for a systematic query of the electronic health records (EHR) to determine whether patients on hydroxychloroquine are less likely to develop PAH.
Eternacept
Although inflammation promotes PAH, the mechanisms by which inflammation and BMPR2 dysfunction conspire to cause disease remains unknown. It was recently shown that the tumor necrosis factor-α (TNFα) selectively reduced BMPR2 transcription and mediated post-translational BMPR2 cleavage via the sheddases, ADAM10 and ADAM17 in pulmonary artery smooth muscle cells. 105 The TNFα inhibitor, Etanercept, was able to reverses disease progression in the Sugen/Hypoxia rat model of experimental PH and restored normal BMP signaling. Eternacept is FDA approved to treat rheumatoid arthritis, juvenile rheumatoid arthritis, psoriatric arthritis, plaque psoriasis and ankylosing spondylitis and is known to be a potent inhibitor of TNFα, the “master regulator” of the inflammatory response in many organ systems. 106 Etanercept is a fusion protein where the TNF receptor is fused to an IgG1 antibody. Given its dual mode of action-targeting the immune system as well as BMPR2 signaling, Etanercept is a promising new repurposed drug that could be easily tested clinically.
Enzastaurin
In an attempt to identify modifier genes that might reduce BMPR2 expression and signaling in PAH and that could be therapeutically targeted, a genetic siRNA HTS screen was combined with an in silico approach of publicly available gene expression data of PAH patients. 46 The tumor suppressor gene, fragile histidine triad (FHIT) was identified as a gene that was both an activator of BMPR2 expression as well as consistently downregulated in blood from PAH patients. The cancer drug enzastaurin, a protein kinase C beta inhibitor that had already been tested in phase I–III lymphoma trials, 107 had been previously reported to increase FHIT. 108 Furthermore, the gene expression signature of enzastaurin resembled, in large parts, the anti-PAH signature, supporting the hypothesis that enzastaurin might be beneficial in PAH. 46 FHIT was reduced in the blood of PAH patients and might be a potential modifier of PAH penetrance in familial PAH as healthy carriers of the same family seemed to have higher FHIT levels than their diseased family members. To support the importance of low FHIT levels in promoting PH, it was shown that FHIT-deficient mice were more prone to develop PH after exposure to chronic hypoxia and that they failed to properly recover in normoxia. Enzastaurin increased FHIT, BMPR2, and ID1 expression in PAECs and was able to improve PH in sugen/hypoxia/normoxia rats by reducing neointima formation, again associated with an increase in FHIT and BMPR2 expression in whole lung tissue. 46 While enzastaurin had shown encouraging preclinical results for the inhibition of proliferation and induction of apoptosis of cancer cells and limited cytotoxicity within phase I clinical lymphoma trials, it showed poor efficacy in phase II and III clinical trials, both in combination with other drugs and as a single agent. 109 Reasons were poor preclinical animal models, inappropriate endpoint analysis, limited standards in phase I clinical trials, as well as insufficient use of biomarker analysis and patient stratification, all factors that would need to be taken into account for a planned future clinical trial in PAH.
Targeting the right ventricle
Carvedilol
The prognosis of those with PAH depends on how the right ventricle responds to the underlying disease. As RV function worsens, symptoms increase and the prognosis becomes worse. 110 In patients with LV failure (heart failure with reduced ejection fraction [HFrEF]), improvement in the LVEF correlates with improvement in patient survival. 111 Carvedilol, a non-selective beta-adrenergic receptor (BAR) antagonist, has been well studied in HFrEF and found to improve patient outcomes such as LVEF, hospitalizations, and mortality. 112
Due to the need to find medications that could help the right ventricle improve its adaptation to PAH, and the success of using carvedilol in the left ventricle, efforts to study carvedilol in PAH were initiated. In a rodent model of PAH, carvedilol was found to be safe and improved both exercise capacity and right ventricular ejection fraction (RVEF). 113 This led to a small pilot study in PAH patients, which also found a significant improvement in RVEF. 114 A subsequent single-center, phase II, RCT of 30 patients (PAHTCH) found that use of carvedilol was well tolerated and was associated with reduction in RV glycolytic rate and increase in BAR levels, both of which may be favorable in helping the right ventricle adapt to PAH. 115 There is currently a phase II study looking to further define the potential effect of carvedilol on the right ventricle (www.clinicaltrials.gov NCT02507011). If successful, carvedilol could be repurposed in clinical use to treat PAH patients with compensated RV failure who are stable on existing therapy. However, unlike other therapies in this review, carvedilol does not seek to impact the underlying pathogenesis that impacts the pulmonary arterioles.
Repurposing medications for pulmonary arterial hypertension: challenges and future directions
The journey from drug discovery to use in clinical practice is long and difficult. As mentioned earlier, repositioning and repurposing both offer an advantage over de novo drug development, as there is less basic science research and less safety data to accumulate. In addition, with efforts for philanthropic drug repositioning, clinical use occurs without FDA approval of a new indication, which offers further time and financial benefit. In repurposing efforts, academic medical centers often play a pivotal role. Participating in drug repurposing offers the academic community several key rewards: improved patient care through new and improved treatments; job creation; the potential for capital revenue through drug research and development; and the dissemination of knowledge and values to the next generations of scientists and providers. 116 In addition, academic medical centers are now including patient care improvements, patient safety, and quality improvements in academic promotion. 117
Despite these advantages, repurposing or repositioning of a medication for use in PAH has a difficult path forward. Because PAH is a relatively uncommon disorder, and there are many existing therapies approved for treatment, the perceived need (by industry and both for and non-profit funding sources) is less when compared to a common disease which lacks treatment (for example, Alzheimer's). This perception of “market saturation” can create difficulty in moving a promising therapy forward to later stages of clinical development.
Late-stage clinical trials in PAH (phase II–III) typically require participation from multiple centers for completion; this significantly raises the financial and human resources required. Millions of dollars are typically required to invest in such a study and successful funding through the NIH is difficult and rarely covers the entirety of the project. Moreover, most academic facilities do not have adequate funding or human resources to support drug discovery programs and the location of an academic center has a significant impact on its ability to secure additional financial assistance from industry. 118 Unless the repositioning or repurposing effort offers the potential for financial return, the manufacturer may not be willing to make the significant financial investment in the effort. However, an effort that does promise financial return would likely do so by introducing an expensive medication to the market. Because the expense of existing medications is one of the current impediments to care for PAH patients, collaboration with industry to provide an optimal PAH therapy creates the conundrum that additional research funding creates higher therapeutic costs. While most investigators have an in-depth understanding of their potential therapy and of PAH, there is a paucity of knowledge and experience regarding the above interplay between funding sources, multiple academic centers, and industry. The expense, time, and knowledge required to successfully complete a late-stage clinical trial are vast, and these impediments to progress are at risk of derailing some or all of the above promising therapies.
Despite the difficult challenges above, there is a path forward for drug repurposing in PAH. There are many promising therapies and many of these therapies offer to combine improved knowledge of disease pathogenesis with identification of biomarkers, to offer new avenues to treatments that directly target underlying mechanisms of disease. Many academic institutions are improving their existing infrastructure to better align investigators with other essential participants in drug repurposing (Blavatnik Biomedical Accelerator of Harvard, the Tri-Institutional Therapeutics Discovery Institute of Memorial Sloan Kettering, the SPARK Translational Research Program of Stanford, etc.) while external organizations have been developed that can help to align investigators without such internal support with both the needed financial and human resources (Cures Within Reach, Biovista, etc.). There are also initiatives in place that can incentivize industry to create therapies for rare diseases such as PAH, including the Priority Review Voucher program in the U.S such as the pediatric rare disease program, given that PAH affects children and adults alike. Through improved collaboration and incentives, there is hope for moving medications through the development process required for repurposing or repositioning a medication.
Conclusion
While many therapies are available for those with PAH, existing therapies do not alter the underlying disease pathogenesis. There is a need for new therapies that will target the underlying mechanisms leading to PAH. As we learn more about genetics, proteomics, and metabolomics in PAH, there is hope of identifying novel biomarkers to optimize patient selection for both research and for therapeutic interventions. Drug repurposing holds promise as a modality to find such novel PAH therapies. Many current therapies under investigation are examples of repurposing, often with associated biomarkers as precision medicine makes its way into the treatment of PAH. The significant challenges to repurposing therapy in PAH may be offset by improving institutional and external support for repurposing and this may hold the key to whether the current promise of repurposing in PAH fulfills its potential to benefit our patients.
Footnotes
Conflict of interest
The authors declare the following conflicts of interest: ES has served as scientific adviser for Selten Pharma, Inc., Vivus (modest). ES is listed as inventor on patent applications Use of FK506 for the Treatment of Pulmonary Arterial Hypertension (Serial No 61/481317) and Enzastaurin and Fragile Histidine Trial (FHIT) Increasing Agents for the Treatment of Pulmonary Hypertension (PCT/US2018/033533). BB serves as a scientific advisor to Healx (Cambridge, UK), Rediscovery Life Sciences, and OneThree Biotech. The other authors declare that there is no conflict of interest.
Funding
This study received the following funding: ES: NHLBI R01 HL128734, Department of Defense grant PR161256, Vera Moulton Wall Center for Pulmonary Vascular Disease Stanford, Pulmonary Hypertension Association (PHA) career development grant, SPARK grant; DG: UL1TR000058 from the National Center for Research Resources, VCU Johnson Center; and AA: 1T32HL129970-O1A1 grant.