
Abstract
Transforming growth factor-β (TGF-β) inhibition is an investigational therapy for pulmonary arterial hypertension with promising results in experimental studies. The present work compared this approach with endothelin-receptor blockade and evaluated the effects of combined administration. Pulmonary arterial hypertension was induced by single monocrotaline injection (60 mg/kg) in 75 Wistar rats and 15 rats served as controls. Intervention groups consisted of treatment with an antibody against TGF-β-ligand, bosentan, both or none, initiated four weeks after monocrotaline injection. Right ventricular systolic pressure, pulmonary vascular remodeling, and exercise tolerance were evaluated eight weeks after monocrotaline injection. Either treatment, alone or in combination, lowered mortality. Comparable efficacy was found in the three treatment groups in terms of right ventricular systolic pressure (~45% decrease) and hypertrophy (~30% decrease), as well as exercise capacity. The three treatment groups equally ameliorated pulmonary vascular remodeling, evidenced by decreased vessel-wall thickness (in vessels 50–200 μm) and a smaller number of pre-∗∗∗capillary arterioles (< 50 μm) with a muscularized media. Treatment either with an antibody against TGF-β or with endothelin receptor blockade are equally effective in experimental pulmonary hypertension. Their combination provides no added benefit, indicating common mechanisms of action.
Keywords
Pulmonary arterial hypertension (PAH) is an infrequent condition, associated with high morbidity and mortality. PAH is caused by remodeling of the pulmonary vasculature and resultant increased resistance, leading to right heart failure.[1] The precise pathophysiological mechanisms of the disease remain incompletely understood, but endothelin-1 (ET-1) and transforming growth factor-beta (TGF-β) have been shown to be critical elements in the pathogenesis of PAH.[2]
ET-1, an endothelium-derived 21-amino-acid-peptide with potent vasoconstrictive and mitogenic actions,[3] has been long recognized as an important mediator of PAH development.[4] The identification of the central role of ET-1 has led to the advent of ET-receptor antagonists that have become a mainstay in the treatment of PAH, after their demonstrated efficacy in improving pulmonary hemodynamics and exercise capacity.[1,5] However, current therapeutic approaches are ineffective in a substantial proportion of patients;[1,6,7] therefore, there is a need for novel treatments, directed at halting the progression of pulmonary vascular remodeling and right ventricular (RV) hypertrophy.
TGF-β is a multifunctional cytokine, regulating the proliferation, differentiation, migration, and survival of various cell types.[8] Based on these actions on the pulmonary vasculature, TGF-β has been implicated in the pathophysiology of PAH, a notion supported by a substantial body of experimental[9] and clinical[10,11] data. As a result, the effects of TGF-β-inhibition have attracted considerable scientific interest. In the in vivo rat model of monocrotaline-induced PAH, treatment with an antibody against TGF-β receptor I improved PAH, RV hypertrophy, and pulmonary vascular remodeling.[12,13] Using the same rat model, our group recently reported attenuated pulmonary vascular remodeling after administration of an antibody against TGF-β ligand, resulting in lower RV systolic pressure and ameliorated RV hypertrophy.[14]
Based on the above considerations, there is evidence that TGF-β inhibition (targeting either the TGF-β ligand[14] or the receptor[12,13]) carries potential therapeutic implications in PAH, but the efficacy of this approach has not been previously compared with ET-receptor blockade. More importantly, it is unknown whether these beneficial effects are exerted over and above ET-receptor blockade and whether combined administration of both treatments produces superior results.
We hypothesized that combined treatment with TGF-β-inhibition and ET-receptor blockade may act synergistically. Therefore, the purpose of the present study was two-fold: (1) To compare the effects of TGF-β-inhibition (by neutralizing TGF-β ligand) with those of ET-receptor blockade; and (2) to examine whether combined treatment with both agents increases the efficacy of each individual therapy. In addition to RV systolic pressure, the end-points of the present study included histological indices of pulmonary vascular remodeling and RV hypertrophy, as well as exercise capacity.
MATERIALS AND METHODS
Experimental animal population
The animal study population consisted of 90 Wistar rats (weighing 377 ± 63 g). All animals received humane care, according to European legislation (European Union Directive for the Protection of Animals Used for Scientific Purposes, 2010/63/EU); they were housed one to two per cage, under optimal laboratory conditions (controlled temperature, humidity, and 12:12h-light:dark cycles), with free access to water and standard rodent chow. The experiments in the submitted work adhere to internationally accepted guidelines for the use of animals in research and the study protocol was approved by the responsible regulatory state authorities (Department of Veterinary Medicine and Animal Welfare, Prefecture of Eastern Attica, Athens, Greece).
The animals were randomly assigned into the following groups: (A) Control group (n = 15); (B) PAH treated with an ET-receptor antagonist (n = 15); (C) PAH treated with an antibody neutralizing TGF-β ligand (n = 15); (D) PAH treated with an antibody neutralizing TGF-β ligand plus an ET-receptor antagonist (n = 15); (E) PAH group (n = 15); and (F) PAH treated with an irrelevant antibody (n = 15). The latter two groups were subsequently examined as a single PAH-control group, as explained below. The study protocol is depicted in Figure 1.
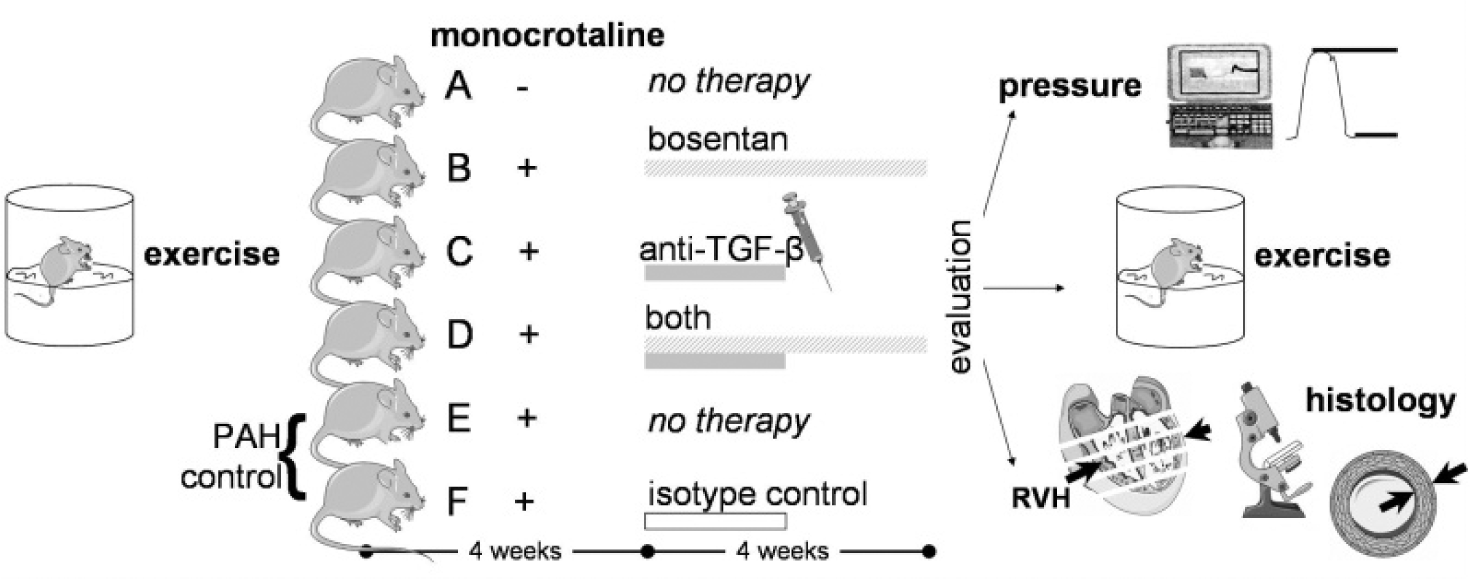
Study protocol.
Induction of PAH
PAH was induced by monocrotaline (Crotaline C2401, Sigma-Aldrich Ltd., St. Louis, Mo., USA), administered as a single subcutaneous injection of 60 mg/kg, as previously published.[15] Monocrotaline pyrrole is activated in the liver and its metabolites cause direct endothelial damage, which, in turn, activates the ET-system and leads to the development of severe PAH.[16] The alterations in molecular signaling and histological changes observed in this model closely resemble human familial or idiopathic PAH.[17]
TGF-β inhibition and ET-receptor antagonism
For TGF-β inhibition, we used a murine monoclonal antibody (1D11, IgG1) that neutralizes all three mammalian isoforms of TGF-β (TGF-β1, TGF-β2 and TGF-β3, often referred to as “pan-specific” antibody) in vitro and in vivo,[18] kindly provided by Genzyme Corporation, Framingham, Massachusetts, USA. Treatment was commenced on Day 28 (after PAH induction) and was delivered intravenously every three days at a dosage of 5 mg/kg for 15 days. As previously noted,[19,23] an isotype-control treatment was given in Group F, using an isotype-matched irrelevant murine IgG1 monoclonal antibody (13C4, 5 mg/kg body-weight), directed against Shigella toxin, also produced by Genzyme. For ET-receptor antagonism, the dual (A and B) ET-receptor antagonist bosentan was used, kindly provided by Actelion Pharmaceuticals Ltd., Allschwil, Switzerland. Fresh preparations were made every day in the form of micro-suspension in 5% Arabic gum, containing bosentan; treatment was commenced on Day 28 (after PAH induction) and was administered orally (300 mg/kg/day) by gavage once daily for 28 days. The dosage of bosentan (300 mg/kg/day) used in our experiments represents the maximal effective dose, being at the plateau of the dose-response curves[24] and has been shown to produce potent and sustained pharmacological effects.[25]
Study end-points
The animals were inspected daily and mortality was recorded. Four weeks after monocrotaline injection, exercise capacity was evaluated, followed by RV pressure recordings and histological assessment.
Exercise capacity
Exercise capacity was assessed with the use of the modified forced-swimming test.[26] To compensate for individual variations in the exercise capacity, the test was performed twice: Once prior to monocrotaline administration, and again at the end of the experiment.
As described previously,[14,26] the animals were placed in a cylinder beaker (height: 50 cm; diameter: 30 cm) filled with water (25°C) to a height of 25 cm. The actual swimming time (measured with a stopwatch) was defined as the total time, i.e., from immersion until near-drowning, from which floating time was subtracted. This test evaluates high-intensity activity and provides a good indicator of aerobic exercise capacity.[26]
RV pressure measurements
Following intubation of the trachea, the animals were mechanically ventilated with the use of a rodent ventilator (model 7025, Ugo Basile, Comerio, Italy) and anesthesia was maintained with isoflurane mixed with oxygen. Open-chest measurements were performed as previously described,[27] after slight modification in our laboratory.[28] Pressure recordings were performed using the Fukuda Denshi/ Datascope (Model IB5006) system.
Histology
Histology was performed by an author (C.G.) blinded to treatment assignment. The animals were sacrificed with potassium chloride, and the heart and lungs were resected en bloc and fixed in neutral-buffered formalin (10%).
RV hypertrophy
A central transversal section of the heart was embedded in paraffin, cut in 2-μm-thick sections and stained with hematoxylin-eosin. RV hypertrophy was expressed as follows: Right ventricular free wall thickness/(left ventricular free wall thickness + interventricular septal thickness)/2.
Pulmonary vascular remodeling
After initial cutting in 2-mm-thick sections, the lungs were embedded in paraffin and were finally cut in 2-μm-thick sections. Lung parenchyma sections were stained with hematoxylin-eosin and immunohistochemically for α-smooth muscle actin (1:100, Dako, Glostrup, Denmark). Pulmonary vascular remodeling was examined by evaluating the following parameters, as described previously.[29,30]
Wall thickness: In 20 (per animal) randomly selected medium-sized pulmonary arteries (with an external diameter 50–200 μm), the external diameter and the medial muscular layer thickness were measured and reported as muscular wall thickness/external diameter
Muscularized arterioles: The number (per optical field) of (normally nonmuscular) precapillary pulmonary arterioles (with an external diameter less than 50 μm) associated with alveolar sacs and ducts. Muscularization was assessed as the number of vessels displaying over 75% of the circumference positive for α-smooth muscle actin in 20 randomly selected fields (using the x100 magnification).
Statistical analysis
Statistical analysis was performed with the use of “Statistica” software package, version 5, (StatSoft Inc., Tulsa, Okla., USA). All values are reported as mean ± standard deviation. Kaplan-Meier survival curves were constructed for each group and heterogeneity was examined by Chi-square. All continuous variables showed a normal distribution, as per Kolmogorov-Smirnov test for normality. Differences between two groups were compared with unpaired Student's t-test and differences between more than two groups were compared with the use of one-way analysis of variance, followed by post-hoc Bonferroni test. Statistical significance was defined at an alpha level of 0.05.
RESULTS
Control group
No difference was found in any variable between the PAH-cohort (Group E) and the PAH-cohort treated with an irrelevant antibody (Group F). Thus, for presentation purposes, these two groups are described as a single PAH-control group (E and F). The five ensuing groups were comparable in terms of body weight (F = 0.67, P = 0.61).
Mortality
Kaplan-Meier survival curves are shown in Figure 2. At the 28th day, mortality rates for groups A, B, C, D, and E and F were 0%, 0%, 0%, 0%, and 50%, respectively, yielding a significant heterogeneity (x2 = 25.3, degrees of freedom = 4, P = 0.00004).
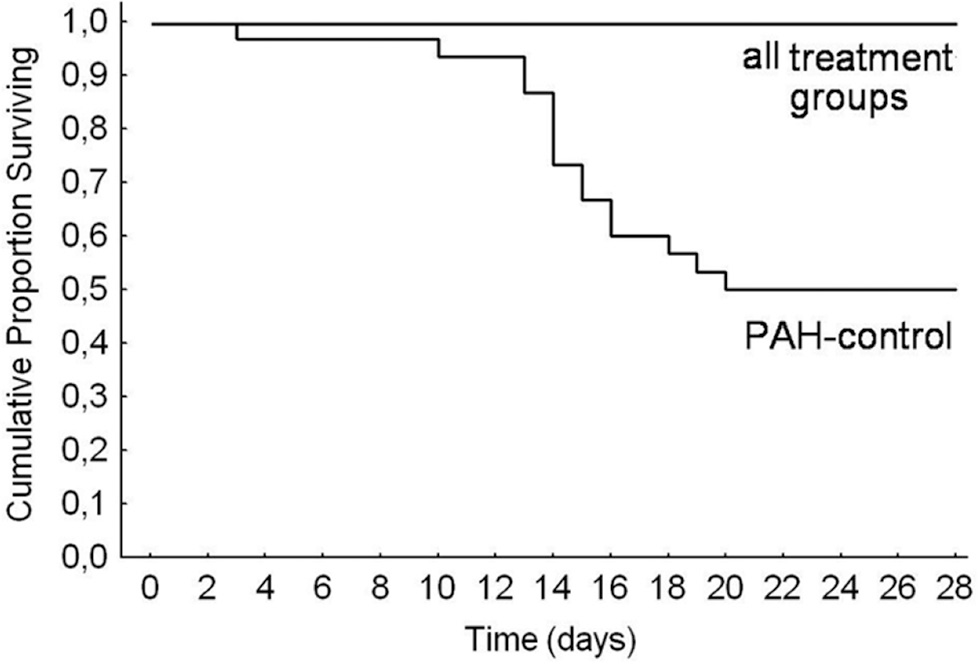
Kaplan-Meier survival curves of the five groups. Mortality rates were 0% in the three treatment groups (bosentan, anti-TGF-β, anti-TGF-β+bosentan), which were lower than the 50% mortality in control rats with pulmonary hypertension.
RV systolic pressure
There was a significant variance in RV systolic pressure in the five groups (F = 25.4, P < 0.0001), as shown in Figure 3. Values were comparable in the three treatment groups (bosentan, anti-TGF-β, anti-TGF-β plus bosentan), in which they were lower (P < 0.001) than the PAH-control group. RV systolic pressure in the anti-TGF-β and anti-TGF-β plus bosentan groups was not significantly different compared to normal controls, but it was higher (P = 0.017) after treatment with bosentan. Representative examples are shown in Figure 4.
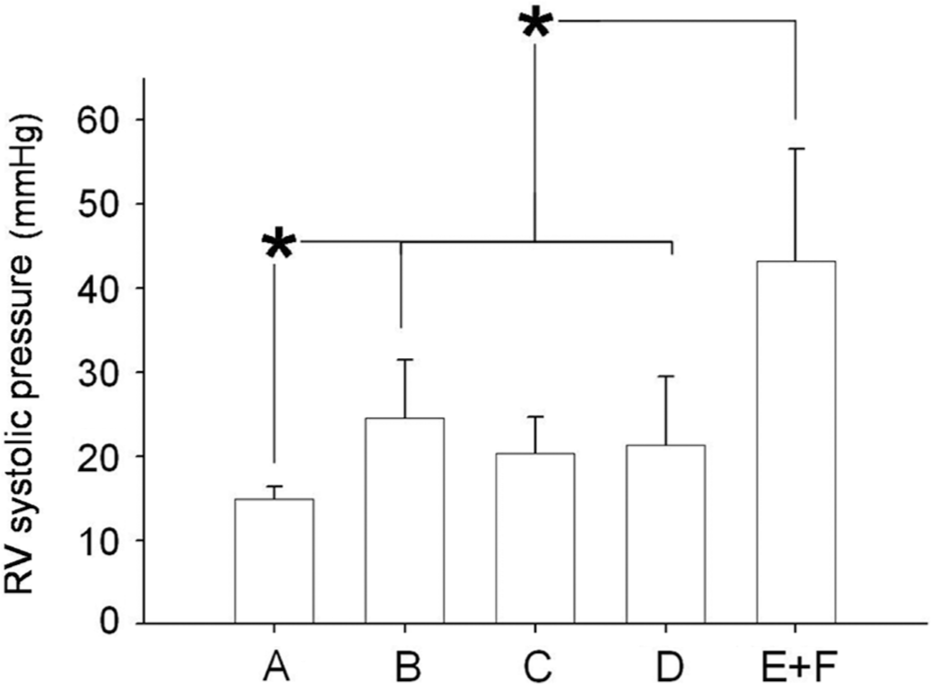
Right ventricular systolic pressure. Right ventricular systolic pressure was comparable in the three treatment groups, in which it was lower than control rats with pulmonary hypertension.
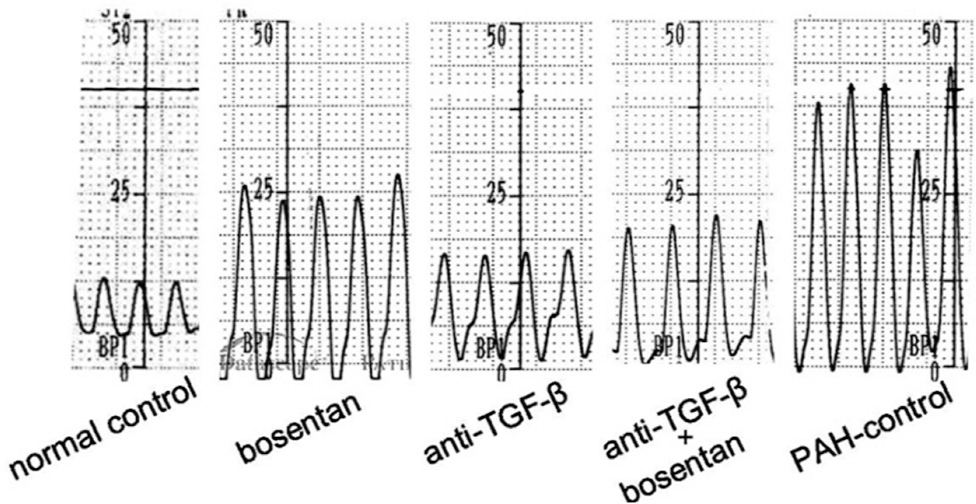
Right ventricular systolic pressure. Representative examples from right ventricular systolic pressure recordings in the five groups.
Exercise tolerance
There was a highly significant variance in the exercise duration (expressed as percent compared to baseline) in the five groups (F = 93.3, P < 0.0001, Fig. 5). Exercise duration was comparable in the three treatment groups, in which it was higher (P < 0.001) than in the PAH-control group. However, exercise duration was lower (all P < 0.001) than in normal controls.
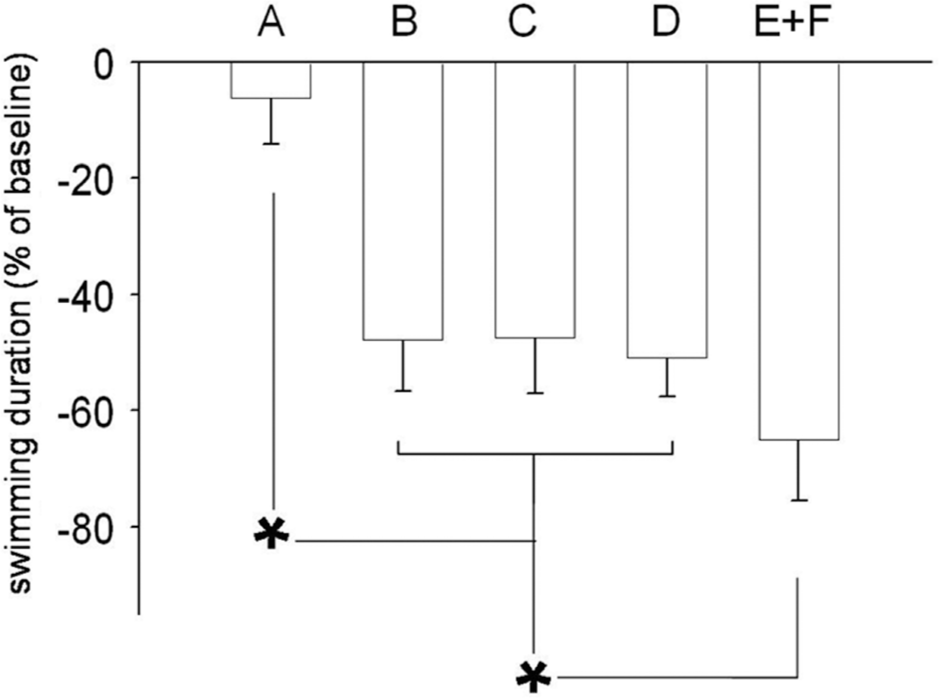
Exercise tolerance. Exercise duration was comparable in the three treatment groups, in which it was higher than in control rats with pulmonary hypertension, but lower than in normal controls.
RV hypertrophy
There was a significant variance in RV hypertrophy (F = 14.8, P < 0.0001) in the five groups, as shown in Figure 6. RV hypertrophy was comparable in the three treatment groups, in which it was lower (all P < 0.01) than that observed in the PAH-control group. Compared to normal controls, RV wall-thickness was higher in the anti-TGF-β group (P = 0.022) and anti-TGF-β plus endothelin receptor antagonist (P = 0.023), whereas it was similar (P = 0.10) in the bosentan group.
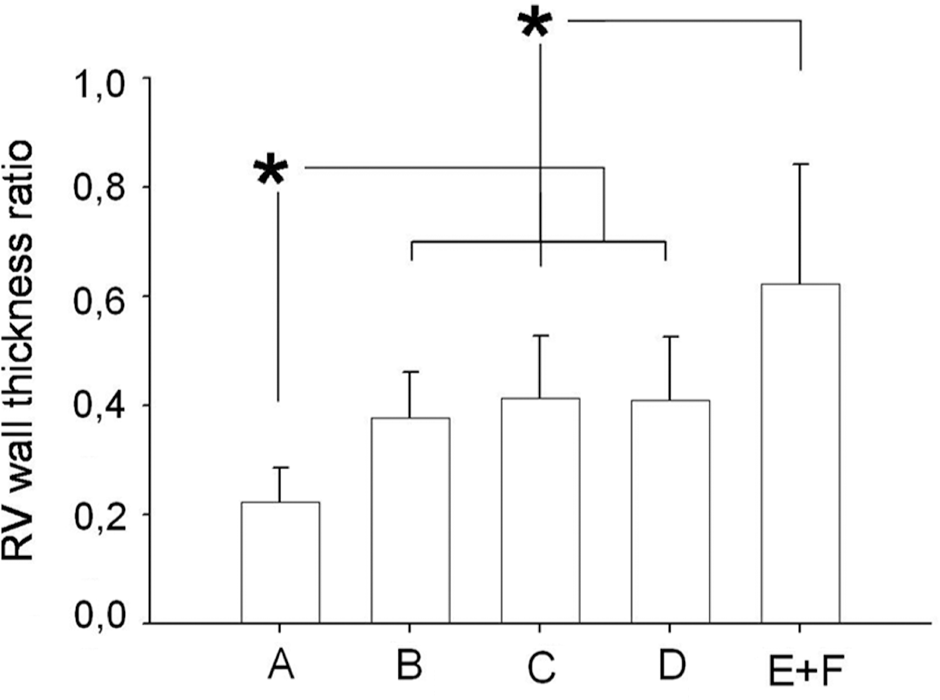
Right ventricular hypertrophy. Right ventricular hypertrophy was similar in the three treatment groups, in which it was lower than in control rats with pulmonary hypertension.
Pulmonary vascular remodeling
A significant variance (F = 49.4, P < 0.0001) was found in wall thickness (corrected for vessel size) in pulmonary vessels with an external diameter ranging from 50 to 200 μm. These values were comparable in the three treatment groups, in which they were lower (all P < 0.00001) than in the PAH-control group (Fig. 7). With the exception of the anti-TGF-β group (P = 0.11), wall thickness was higher in the bosentan (P = 0.047) and in the anti-TGF-β plus bosentan (P = 0.00048) groups than in normal controls. Representative examples are shown in Figure 8.
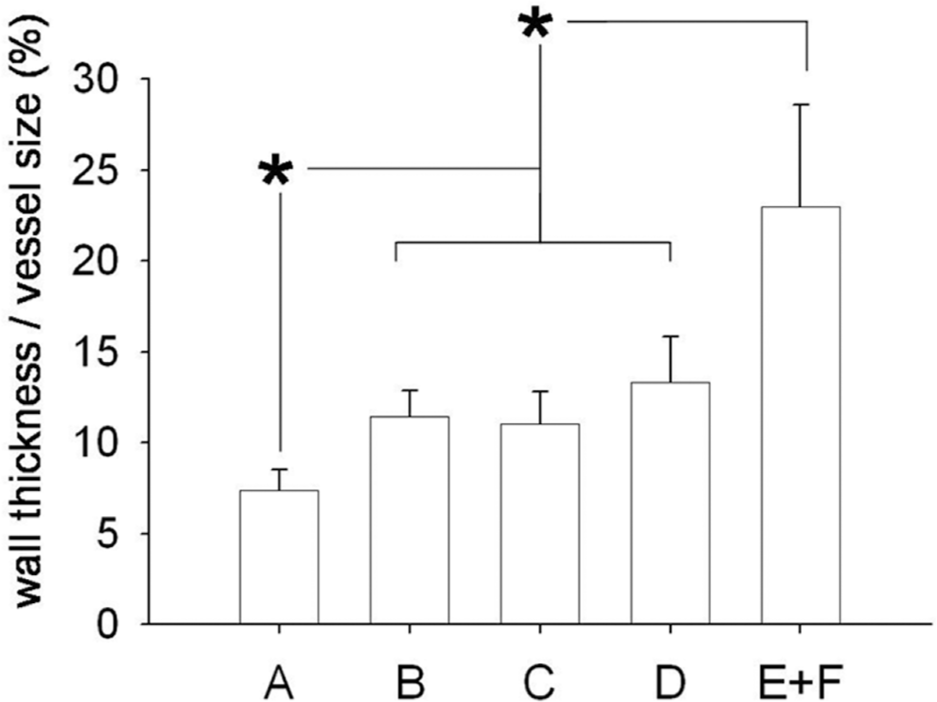
Wall thickness in medium-sized pulmonary arteries. Wall thickness (corrected for vessel size) was similar in the three treatment groups, in which it was lower than in control rats with pulmonary hypertension.
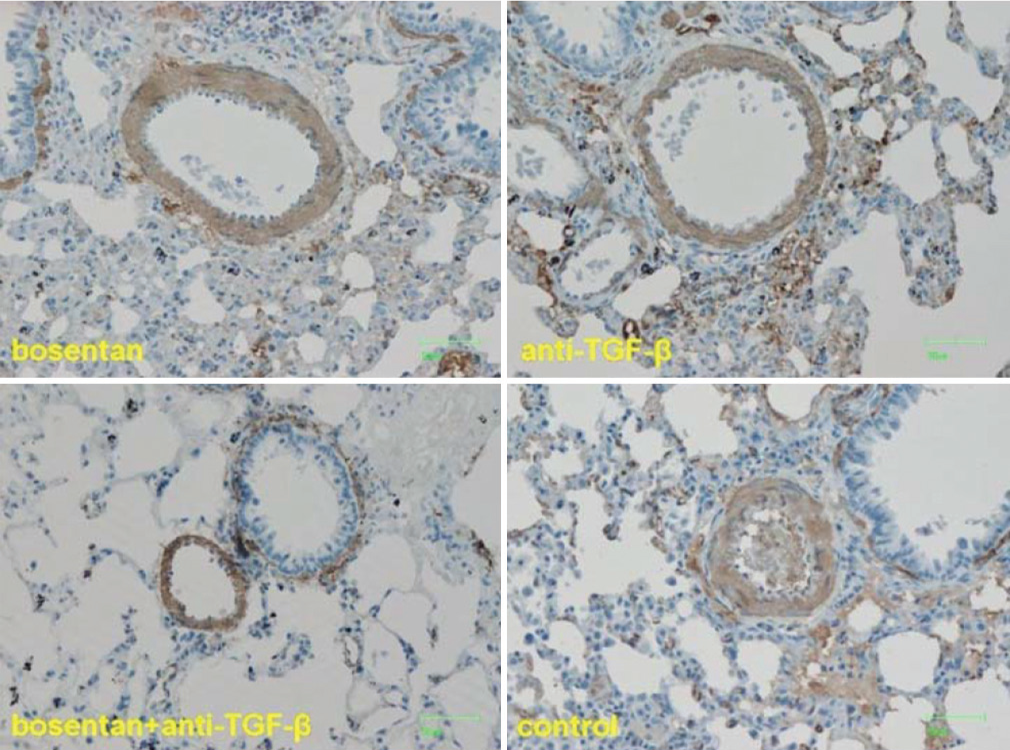
Wall thickness in medium-sized pulmonary arteries. Representative examples from the three treatment groups and control rats with pulmonary hypertension.
There was also a significant variance (F = 56.4, P < 0.0001) in the number of precapillary arterioles (with external diameter less than 50 μm) displaying a muscularized media, as shown in Figure 9. Values were comparable in the three treatment groups, in which they were lower (all P < 0.00001) than in the PAH-control group, but higher (all P < 0.025) than in controls.
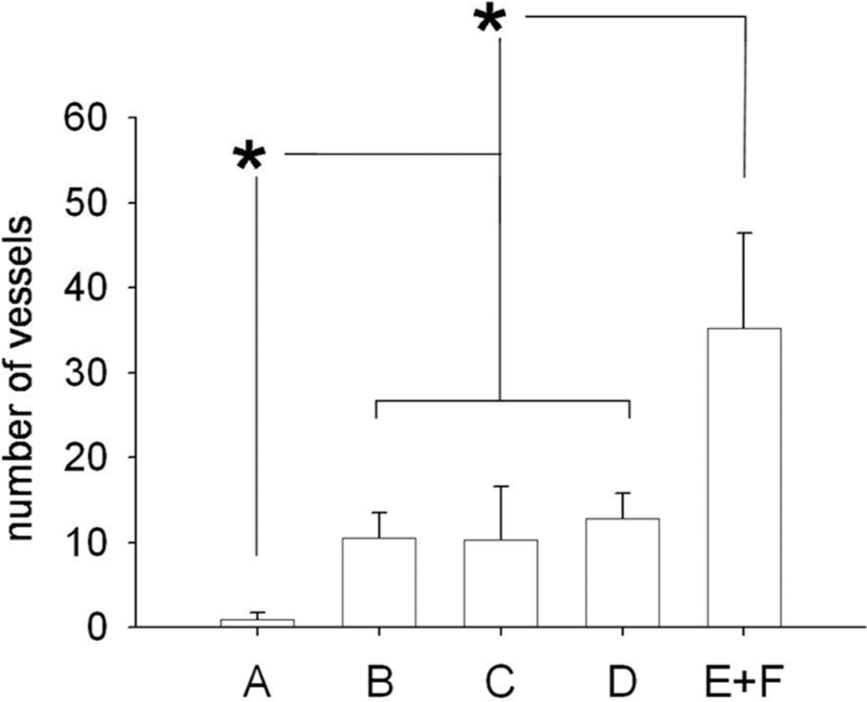
Muscularized pulmonary arterioles. The number (per optical field) of muscularized pulmonary arterioles was similar in the three treatment groups, in which it was lower than in control rats with pulmonary hypertension.
DISCUSSION
Comparison between anti-TGF-β-antibody and bosentan
The present study compared the effects of TGF-β inhibition (by neutralizing TGF-β ligand) and ET-receptor blockade in rats with monocrotaline-induced PAH. Treatment with either anti-TGF-β-antibody or bosentan blunted the increasein RV systolic pressure and the development of RV hypertrophy and attenuated the deterioration of exercise tolerance. These favorable effects were due to ameliorated pulmonary vascular remodeling, evidenced by decreased wall thickness in medium-sized pulmonary arterioles and a reduction in the number of small arterioles displaying a muscularized wall.
TGF-β inhibition in PAH
The effective attenuation of PAH after treatment with TGF-β-inhibition, reported in the present study, is in line with previous studies[12–14] and provides further support on the pathogenetic role of increased TGF-β activity.[9–11] In the previous[14] and present studies from our laboratory, TGF-β activity was neutralized with the use of an antibody against TGF-β ligand, an intervention that has not been examined previously in experimental PAH. In the present work, this treatment decreased RV peak systolic pressure by approximately 45% and ameliorated RV hypertrophy by approximately 30%.
In contrast to neutralizing TGF-β activity, two previous experimental studies[12,13] followed a different TGF-β-inhibition approach, using molecules directed against TGF-β receptor I. More specifically, intraperitoneal administration of a molecule directed against activin receptor-like kinase-5 in vivo inhibited TGF-β/Smad3 signaling and prevented the progression of PAH in the monocrotaline rat-model,[13] evidenced by a 22.3% reduction in RV peak systolic pressure and a 23.5% reduction in RV hypertrophy. Similar results were obtained with an orally active compound,[12] consisting of a 15% reduction in mean pulmonary artery pressure and a 26% increase in cardiac output, accompanied by improved RV diastolic and systolic function indices. Thus, both anti-TGF-β-inhibition approaches appear effective, but should be compared head-to-head in future studies.
Despite the similar intervention in our previous[14] and present experiments, two different “pan-specific” antibodies (which neutralize all three isoforms of TGF-β) were used. In contrast to the less well-studied molecule used previously,[14] the efficacy of the compound used in the present experiments (1D11, Genzyme Corporation) has been demonstrated in vitro and in vivo in a variety of experimental settings, such as in the field of nephrology[20–21] and in cancer research.[23]
Our previous histological analysis[14] revealed that the benefit of TGF-β-inhibition in pulmonary vascular remodeling was confined only in small (<50 μm) arterioles. Here, we further observed a decrease in wall thickness of medium-sized (50-200 μm) pulmonary arterioles. In addition to the different compounds used (as outlined above), the more pronounced attenuation of pulmonary vascular remodeling in the present work can be attributed to three differences: (1) The route of administration; (2) the time point of treatment initiation; and (3) the duration of administration. These points are briefly discussed below.
In contrast to the continuous intraperitoneal administration (via an osmotic mini-pump) in our previous work,[14] as well as in other in vivo studies using the same 1D11-Genzyme antibody,[20–23] intravenous administration was favored in the present work. This choice was based on early information, indicating effective suppression of TGF-β activity after intravenous antibody administration[31] and these findings are consistent with the experience at Genzyme laboratories.[32] Furthermore, unpublished observations suggest that bioavailability may be higher with intravenous than with intraperitoneal administration (Patrick Finn, PhD, Genzyme Corporation, personal communication). Despite these considerations, the optimal route of administration remains to be examined
Previous studies[13] characterizing the rat model of monocrotaline-induced PAH indicated that pulmonary artery pressure and RV hypertrophy are significantly abnormal on Day 7 onwards after monocrotaline-injection, and pulmonary vascular remodeling is largely complete by four weeks. As intervention was commenced at early stages (i.e., seven days post monocrotaline injection) in our previous experiments,[14] the effects of TGF-β-inhibition on pulmonary vessels could not be fully revealed, due to relatively minor changes at this point. In the clinical setting, these findings may apply to the prevention of progression of milder forms of PAH. This differs from the time-point chosen in the present study (i.e., four weeks post-monocrotaline injection), corresponding to a more complex therapeutic strategy. Our present findings concur with those in prior reports,[12,13] where TGF-β-inhibition (using molecules directed against TGF-β receptor I) was initiated three weeks after monocrotaline injection; in this context, the results presented here, examined together with previous findings,[12,13] indicate that holding the progression of advanced pulmonary vascular remodeling is feasible, an observation with potential ramifications in the treatment of patients with severe PAH. However, neither the present, nor previous[12,13] studies reported findings indicative of reversion of established severe experimental PAH
The duration of TGF-β-inhibition may be an important parameter that determines the extent of pulmonary vascular remodeling. In contrast to a 7-day treatment in our previous study,[14] the duration of this course was doubled in the present work. As the mechanisms of medial hypertrophy differ across the pulmonary vascular tree, due to heterogeneity in cellular responses,[17,33–35] our results indicate that more sustained inhibition of growth stimuli is required for attenuation of wall thickening in medium-sized arteries, as opposed to small, precapillary arterioles. Nevertheless, the pathophysiology of remodeling across the pulmonary vascular tree is not completely understood, and further investigation is necessary.
ET-receptor blockade and combination treatment in PAH
The ET-system is markedly activated in PAH, causing vasoconstriction of the vascular endothelium; in addition, ET-1 acts also on smooth muscle cells and fibroblasts on the pulmonary vessel wall, resulting in chronic vascular remodeling.[36] In agreement with previous studies using the same rat model,[37,38] we report effective attenuation of PAH, decreased RV hypertrophy, and improved survival after four-week oral treatment with the dual (ETA and ETB) ET-receptor antagonist bosentan. These data have been confirmed in clinical studies, rendering ET-receptor blockade an established treatment for PAH, used as first-line approach in many subsets of patients.[1,5–7] Despite these considerations, there is often the need for combined treatment with agents with different mechanisms of action,[1,6,7]due to inadequate responses after monotherapy.
The issue of combination therapy in PAH has been actively investigated in both experimental and clinical studies. For example, combination treatment with bosentan and sildenafil attenuated the increase in mean pulmonary arterial pressure and decreased RV hypertrophy more effectively than each therapy alone,[24] and this combination improved RV energetics.[39] Despite the widespread clinical use of combination therapy in many PAH centers, several questions regarding the efficacy of such regimens remain open, as highlighted in current treatment guidelines.[7]
Anti-TGF-β and bosentan
Our study evaluated the effects of combined anti-TGF-β-inhibition and ET-receptor-antagonism, a combination that has not been previously examined. The main finding of the present study was the lack of significant differences in the combination treatment, compared to either therapy alone; in this respect, equal efficacy was found in all end-points examined, namely in RV systolic pressure and hypertrophy, in exercise capacity, and in the histological indices of pulmonary vascular remodeling. These results refute our initial hypothesis on the possible additive effects of combined treatment with TGF-β inhibition plus ET-receptor antagonism and point toward a common mechanism of action of these therapies. This notion is based on a number of experimental data, briefly summarized below.
Prior data from in vitro studies indicate that TGF-β regulates ET-1 synthesis. Markewitz et al.[40] found that TGF-β increased the expression of preproET-1-mRNA and ET-1 production in rat and human pulmonary arterial smooth muscle cells. Likewise, Star et al.[41] found that TGF-β administration (at a wide dose-range) increased ET-1 in human endothelial cells (derived from the vessel wall of small-sized pulmonary arterioles), an action mediated by phosphorylation of the Smad-pathways. A similar effect was shown in human vascular smooth muscle cells, where both bosentan and TGF-β receptor antagonism blunted the increase in cytosolic phosphor-Smad2C and its downstream signaling.[42]
Many actions of TGF-β appear to be mediated by ET-1 through both Smad-independent and Smad-dependent mechanisms. In lung fibroblasts, TGF-β was shown to increase ET-1 production through TGF-β/ALK5- and Jun N-terminal kinase-dependent mechanisms.[43] In the same setting, the notion of ET-1 being a downstream mediator of TGF-β responses was reinforced by the effects of bosentan, which decreased profibrotic markers in response to TGF-β.[44] Lastly, in transgenic mice over-expressing TGF-β, increased ET-1-induced contraction was reported in cerebral arteries, mediated by ETA and ETB receptors.[45]
Treatment effects on exercise capacity
The assessment of exercise capacity in the present work is a valuable parameter because it reflects the functional significance of PAH and evaluates an important aspect of treatment, invariably used as an end-point in clinical studies. Thus, the inclusion of this parameter facilitates the interpretation of the results of experimental studies in the clinical perspective. The comparable exercise tolerance between the three treatment groups (bosentan, anti-TGF-β, anti-TGF-β plus bosentan) reported here is in agreement with our hemodynamic and histological findings; taken together, our findings reinforce our conclusions regarding the equivalent efficacy of the two therapies examined and the lack of added value when used in combination.
A striking difference between the results of our present and previous[14] studies is the magnitude of exercise capacity; in contrast to the previous impressive improvement (threefold longer exercise time over PAH-controls) in anti-TGF-β-treated rats, only a modest (~ 25%) effect was noted in the present experiments. We believe that this diversity should be attributed to the different time points of treatment initiation, representing the therapeutic concepts of PAH prevention versus halting its progression at advanced stages.
Limitations of the study
We feel that the present experimental work may advance current understanding on the pathophysiology of PAH. However, two limitations should be acknowledged:
(1) We did not measure TGF-β levels in the plasma or in the lungs in any animal group; and (2) Perhaps more importantly, our study lacks direct molecular insights on the potential treatment mechanisms as we did not examine the downstream pathways of TGF-β-inhibition
In conclusion, the present study demonstrates that TGF-β-inhibition (by neutralizing TGF-β ligand) and ET-receptor blockade with bosentan equally attenuate PAH when administered late in the course of experimental PAH. Either treatment improved pulmonary vascular remodeling and RV hypertrophy, resulting in enhanced exercise tolerance in the in vivo monocrotaline-rat model. However, combined treatment was not associated with added benefit, implicating common mechanisms of action, a hypothesis that merits investigation in future studies.
Footnotes
ACKNOWLEDGMENTS
Eleftheria Karabela, RN, and Christos Stefopoulos, MD, assisted during the experiments. Eleni V. Goga, MSc, coordinated this research. We are indebted to Genzyme Corporation, Framingham, Massachusetts, USA for providing 1D11 and 13C4 antibodies and to Actelion Pharmaceuticals Ltd., Allschwil, Switzerland for providing bosentan.