
Abstract
Patients affected by pulmonary arterial hypertension (PAH) show a typical pattern of abnormalities on cardiopulmonary exercise testing (CPET). However, CPET is not routinely used as a screening method. We discuss a patient with hereditary PAH in whom CPET revealed onset of disease. Furthermore, we show that the abnormalities observed can improve in part by PAH-specific treatment.
A diagnosis of PAH requires a right heart catheterization, confirming a mean pulmonary artery pressure (mPAP) ≥ 25mmHg and a pulmonary artery wedge pressure ≤ 15mmHg. Echocardiography is the recommended screening method for patient at risk for developing PAH and is frequently used to identify patients requiring right heart catheterization. CPET may also have a role in screening, as the test shows a typical gas exchange pattern in PAH. Here we discuss the case of a patient with hereditary PAH in whom CPET revealed the onset of disease and also reflected the subsequent response to treatment.
CASE REPORT
A 24-year-old female was analyzed for suspected pulmonary arterial hypertension (PAH) because of a prior diagnosis of hereditary PAH (with a BMPR2-gene mutation) in her aunt. At the first screening visit in 2009, she had no physical complaints and no signs of pulmonary hypertension. Furthermore, cardiopulmonary exercise testing (CPET) was normal (Table 1 and Fig. 1).
Results of three consecutive cardiopulmonary exercise tests in a female with hereditary PAH
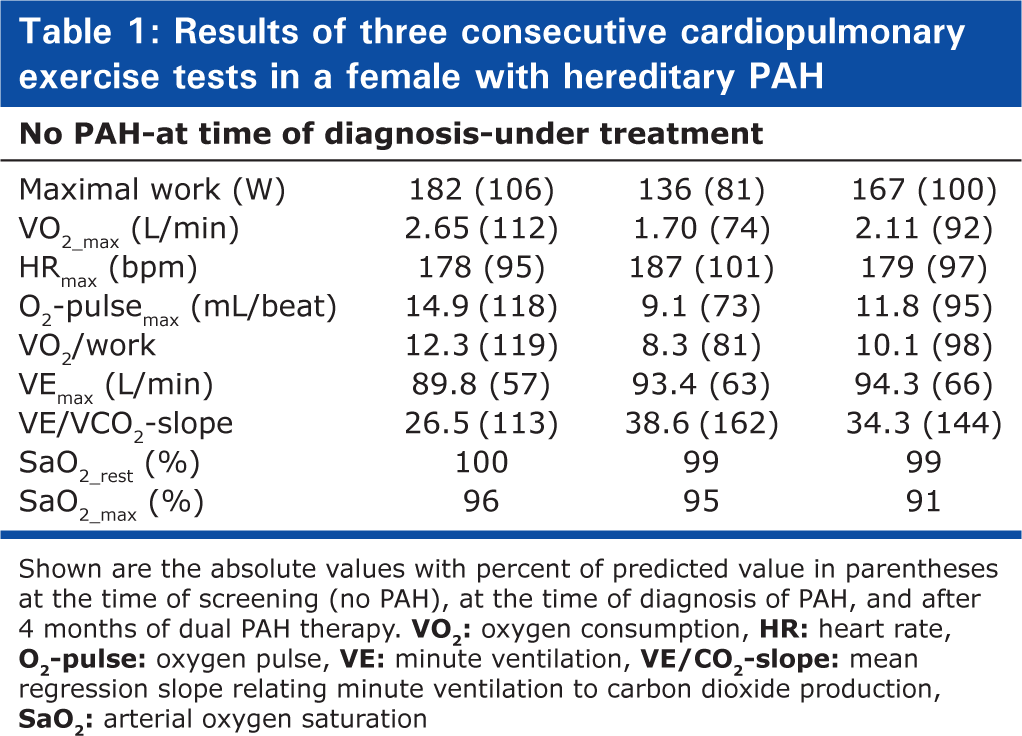
Shown are the absolute values with percent of predicted value in parentheses at the time of screening (no PAH), at the time of diagnosis of PAH, and after 4 months of dual PAH therapy.
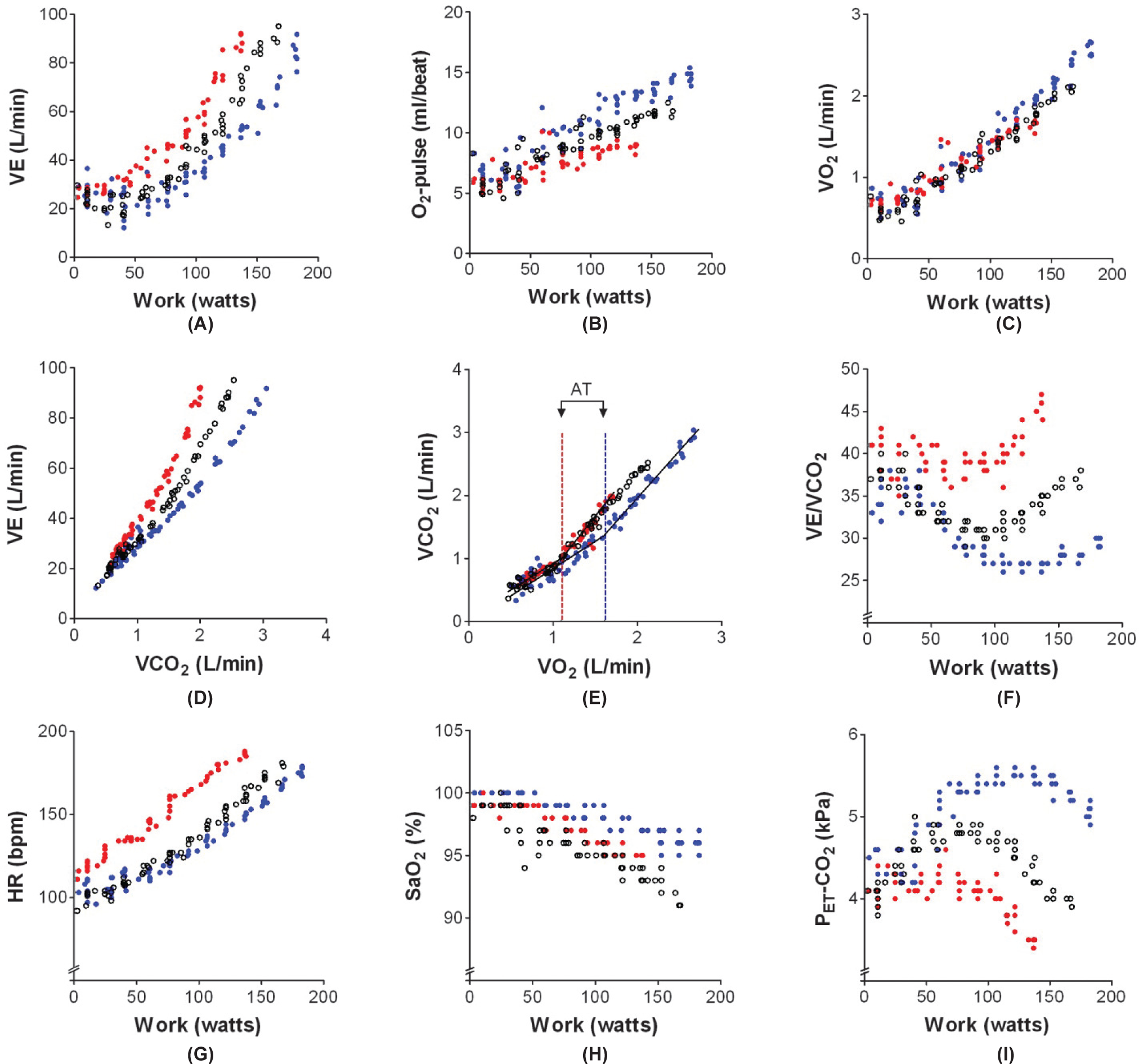
Graphic depiction of the results of three consecutive cardiopulmonary exercise tests (CPET) in a female subject with hereditary pulmonary arterial hypertension (overlay plot, blue dots show CPET results in 2009 when pulmonary hypertension was not present, red dots show the results in 2012 when a diagnosis of hereditary pulmonary arterial hypertension was made, and white dots show CPET results after 4 months of dual PAH therapy).
In 2012, the patient was seen again, now with complaints of a deteriorated physical performance. Echocardiography showed signs of pulmonary hypertension with a moderate increase in right atrial and ventricular size, a tricuspid regurgitation velocity of 3.7 m/s, and an estimated systolic pulmonary artery pressure of 56 mmHg. Right ventricular systolic function was normal and a septal shift was not seen. Due to the high suspicion of PAH, a right heart catheterization was performed. Because of a mean pulmonary artery pressure (mPAP) of 45 mmHg, a cardiac output of 5.0 L/min, a pulmonary vascular resistance of 624 dyn-s-cm−5, and the finding of a BMPR2-gene mutation, a diagnosis was made of hereditary PAH. Laboratory testing revealed a moderately increased NT-proBNP (351 ng/L) and cardiac MRI showed a normal right ventricular end-diastolic volume (158 mL), and a moderately-to-severely reduced right ventricular ejection fraction of 37%.[1,2] Unlike the initial test in 2009, CPET showed a pattern typical for pulmonary hypertension,[3–5] with a low peak oxygen consumption (VO2), oxygen-pulse (O2-pulse), and anaerobic threshold (AT), all indicating a limitation to increase cardiac output (Fig. 1). An increased minute ventilation (VE) for a given workload and for a given volume of carbon dioxide production (VE/VCO2-slope) and a decreased end-tidal CO2 tension (PET-CO2) suggested increased dead space ventilation and perhaps an altered PaCO2 set-point.
Dual therapy with bosentan and tadalafil was started and the patient was re-evaluated four months after the start of therapy. Right heart catheterization was repeated and demonstrated a hemodynamic improvement with a mPAP of 34 mmHg, a cardiac output of 7.0 L/min, and a pulmonary vascular resistance of 285 dyn-s-cm−5. The hemodynamic improvement was accompanied by a normalization of NT-proBNP levels (48 ng/L) and an improved right ventricular ejection fraction (50%). CPET showed an improvement in maximal work, peak oxygen consumption, and maximal oxygen pulse (Fig. 1). In addition, ventilation for a given volume of carbon dioxide production decreased toward normal values and the end-tidal CO2 tension tended to normalize. Remarkably, despite these improvements in resting hemodynamics and exertional gas exchange parameters, arterial oxygen saturation during exercise decreased further to 91% at the end of exercise. We speculate that PAH-specific treatment in this patient, despite lowering PVR, had no favorable effect on the matching of ventilation and perfusion. Perhaps by improving cardiac output without structurally changing remodelled small pulmonary vessels, PAH-specific therapy may further decrease the transit time of erythrocytes within the pulmonary circulation during exercise and thereby worsen oxygen diffusion.
Here we demonstrate that the presence of pulmonary hypertension is characterized by a typical gas exchange pattern during CPET which can improve in part by PAH-specific treatment. Although CPET is not routinely used to screen for the development of PAH in mutation carriers among members of families with hereditary PAH, our findings do suggest that CPET may be a useful screening tool.