
Abstract
A growing body of evidence suggests that exercise pulmonary hypertension (ePH) is an early form of pulmonary arterial hypertension (PAH). Identifying the disease at an early, potentially more responsive phase, and initiating treatment may improve functional status and prevent progression to severe forms of PAH. This was a single-center, open-label six-month treatment trial to evaluate the effect of ambrisentan on pulmonary hemodynamics and exercise capacity in ePH utilizing invasive cardiopulmonary exercise testing (iCPET). After six months of treatment with ambrisentan, patients repeated iCPET; exercise capacity, symptoms, and pulmonary hemodynamics were reassessed. Twenty-two of 30 patients completed the treatment phase and repeat iCPET. After six months of treatment there was a significant decline in peak exercise mPAP (−5.2 ± 5.6 mmHg, P = 0.001), TPG (−7.1 ± 8.0 mmHg, P = 0.001), PVR (−0.9 ± 0.7 Woods units, P = 0.0002), and Ca-vO2 (−1.8 ± 2.3 mL/dL, P = 0.0002), with significant increases in peak PCWP (+2.9 ± 5.6 mmHg, P = 0.02), PVC (+0.8 ± 1.4 mL/mmHg, P = 0.03), and CO (+2.3 ± 1.4 L/min, P = 0.0001). A trend toward increased VO2max (+4.4 ± 2.6% predicted, P = 0.07) was observed. In addition, there were improvements in 6MWD and WHO FC after 24 weeks. Our findings suggest that treatment of ePH with ambrisentan results in improved pulmonary hemodynamics and functional status over a six-month period. Treatment of ePH may prevent the progression of vascular remodeling and development of established PAH.
Introduction
Exercise pulmonary hypertension (ePH) remains a controversial topic among experts in pulmonary vascular disease, and was omitted from the clinical classification of pulmonary hypertension in 2008 1 and 2013 2 due to a lack of universally accepted upper limits of normal for exercise pulmonary hemodynamics. We have previously characterized ePH in a large group of symptomatic patients using direct measurements of pulmonary hemodynamics at rest and during maximum incremental cardiopulmonary exercise testing. 3 The pattern and severity of the pulmonary hemodynamic response in ePH is intermediate between that of normal participants and patients with resting pulmonary arterial hypertension (PAH), 3 and is associated with reduced exercise capacity and decreased quality of life. 4 Similar to what is seen in World Health Organization (WHO) Group-1 PAH, patients with ePH have right ventricular-pulmonary vascular uncoupling 3 and a metabolic profile consistent with a nitric oxide deficient state. 5 Recent studies further support the hypothesis that ePH represents a mild, early phase of PAH.6–10
While significant strides have been made in the development of treatment of PAH, prognosis remains poor. 11 On average, patients are symptomatic for more than two years before a diagnosis of PH is made. 12 By the time of diagnosis, there is already extensive remodeling of the pulmonary vascular bed. As studies have demonstrated, borderline resting or elevated pulmonary arterial pressures during exercise may predict the development of PAH.13–18 Consequently, screening and early detection of ePH might identify a group of patients more responsive to treatment aimed at preventing progression to resting PAH. Few studies have investigated the treatment of ePH patients.19–22 With such a paucity of data, additional studies of treatment of ePH are warranted.
Ambrisentan is a propanoic acid class, A-selective, high affinity endothelin receptor antagonist (ERA) that has demonstrated significant improvement in six-minute walk distance (6MWD) in Phase 2 and Phase 3 efficacy studies in patients with PAH. 23 Clinically meaningful improvements were also seen for Borg Dyspnea Score, WHO functional class (FC), quality of life, and cardiopulmonary hemodynamics, with a low incidence of relevant serum aminotransferase abnormalities.23,24 In addition, the drug’s half-life allows for once-daily dosing, a characteristic associated with improved participant compliance.
We sought to evaluate the effects of ambrisentan in patients with ePH administered orally for six months on pulmonary hemodynamics and exercise capacity utilizing invasive cardiopulmonary exercise testing (iCPET). Secondary objectives include the effect of ambrisentan treatment on improvements in 6MWD and WHO FC.
Methods
Design and study population
This was a single-center, open-label, uncontrolled treatment trial of of ePH with the pulmonary vasodilator ambrisentan. We identified 30 consecutive adults with a newly confirmed diagnosis of clinically stable ePH, not previously treated with any pulmonary vasodilator, and without any clinically significant co-morbidities (see online supplement for full details of inclusion/exclusion criteria and study protocol). Throughout the six-month treatment phase, no patient was treated with another pulmonary vasodilator, enrolled in cardiac or pulmonary rehabilitation, or a formal exercise training program of any kind.
Partners Human Research Committee approved this study (protocol 2008P000687, NCT01338636). All patients were recruited after having had a clinically indicated iCPET performed for the purpose of evaluating unexplained exertional intolerance. Patients were considered for the study if they were aged over 18 years, had findings of ePH on an iCPET performed within the six months prior to entry into the study. Participants provided written informed consent prior to any study-related procedures or assessments. ePH was defined as mean pulmonary artery pressure (mPAP) of > 30 mmHg, pulmonary capillary wedge pressure (PCWP) of < 20 mmHg, and pulmonary vascular resistance (PVR) of > 1 Woods units (WU) at peak exercise and in the absence of resting PAH. 3 Female participants of childbearing potential were required to use a minimum of two forms of contraceptive therapy, including at least one barrier method. In all cases, concomitant medications were stable for at least four weeks prior to enrollment in the study and did not change during the study period and follow-up. Changes in diuretic therapy were made as needed during the study period.
Once enrolled, baseline WHO FC, 6MWD, and Borg Dyspnea Score were assessed. Immediately after all baseline measurements were obtained, ambrisentan therapy was initiated at 5 mg by mouth daily. All patients were evaluated at weeks 4, 8, 12, 16, and 20, during which 6MWD and Borg Dyspnea Score, WHO FC, concomitant medications, and adverse events were assessed. At the week 4 visit, patients tolerating the 5 mg dose were increased to 10 mg for the duration of the study. At the end of the six-month treatment phase, participants underwent a repeat iCPET.
The iCPET methodology has been described elsewhere.25,26 Briefly, simultaneous measurements of ventilation, breath-by-breath respiratory gas exchange, arterial and mixed venous blood gas sampling, and pulmonary hemodynamics are assessed at rest and during incremental upright cycling to exhaustion.
Statistical analysis
Continuous variables are expressed as mean ± standard deviation, while categorical variables are reported as the percentage of patients. Data from the treatment phase were compared to baseline for all efficacy endpoints. If the variable was normally distributed, a paired t-test was used to compare values; the Wilcoxon signed rank test was used for non-normally distributed data. All statistical analyses were performed using Stata 14 (StataCorp LP, College Station, TX, USA) and SPSS 23 (IBM Corp, Armonk, NY, USA).
Results
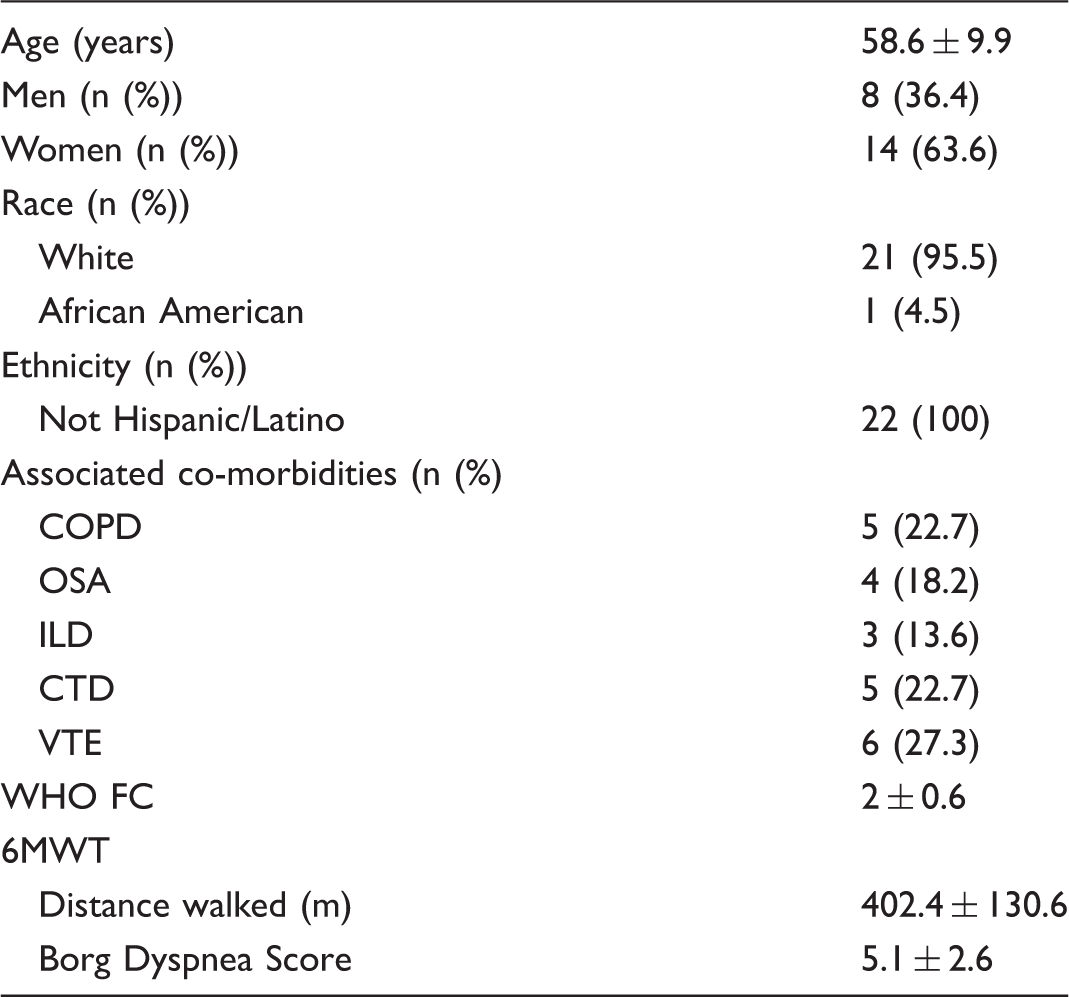
Baseline characteristics of study participants (n = 22).
Baseline characteristics of participants withdrawn (n = 8).
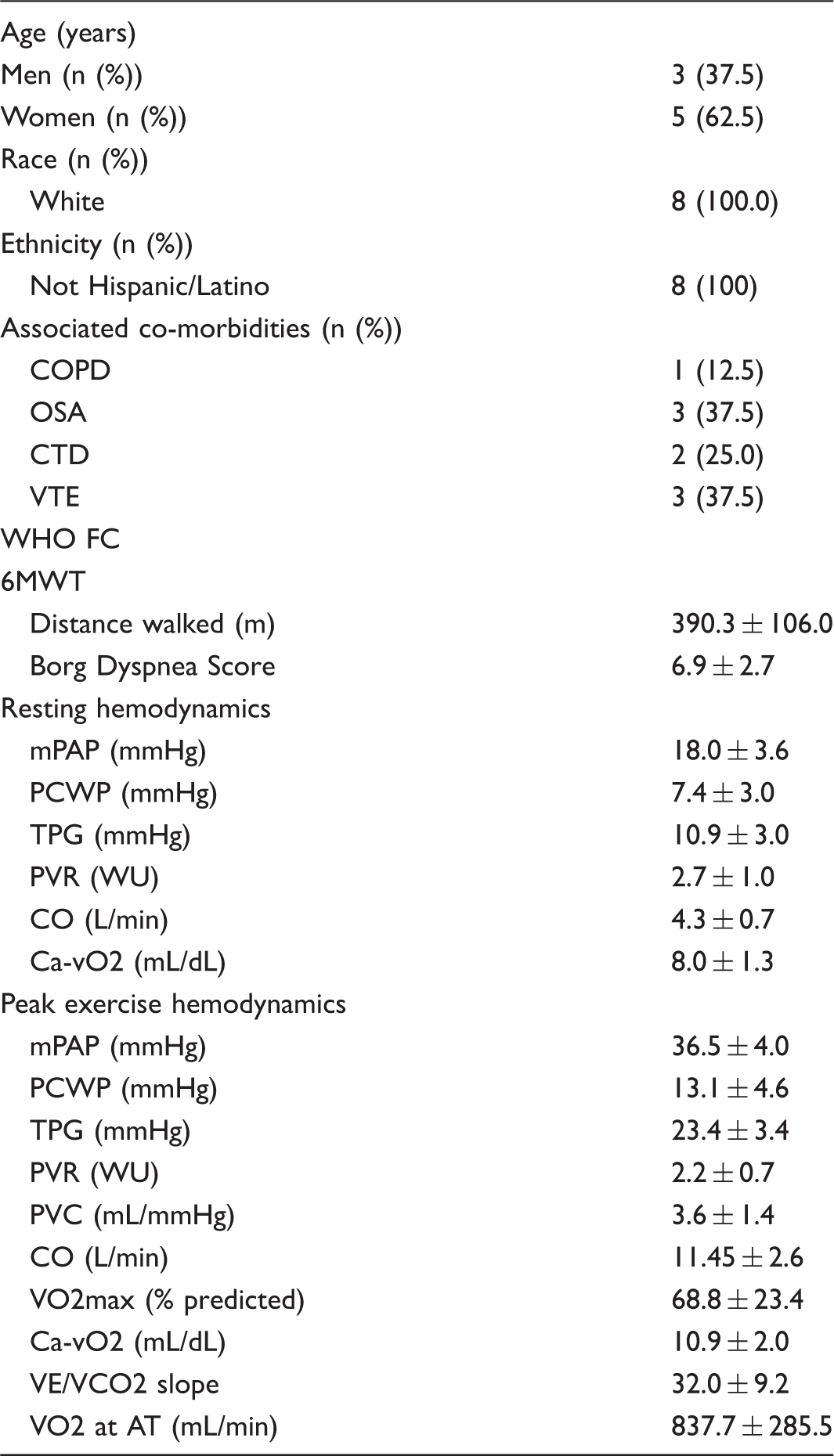
WHO FC, World Health Organization functional class; 6MWT, six-minute walk test; mPAP, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; TPG, transpulmonary pressure gradient; PVR, pulmonary vascular resistance; PVC, pulmonary vascular compliance; CO, cardiac output; VO2max, maximum oxygen uptake; Ca-vO2, arteriovenous oxygen content difference; VE/VCO2, minute ventilation/carbon dioxide production; VO2 at AT, oxygen consumption at anaerobic threshold.
Resting hemodynamics (n = 22).
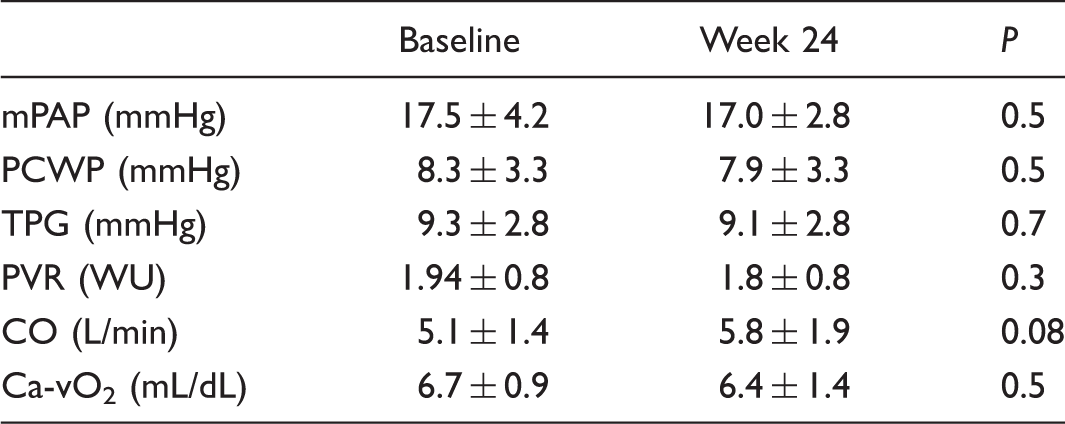
mPAP, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; TPG, transpulmonary pressure gradient; PVR, pulmonary vascular resistance; CO, cardiac output; Ca-vO2, arteriovenous oxygen content difference.
Peak exercise hemodynamics (n = 22).
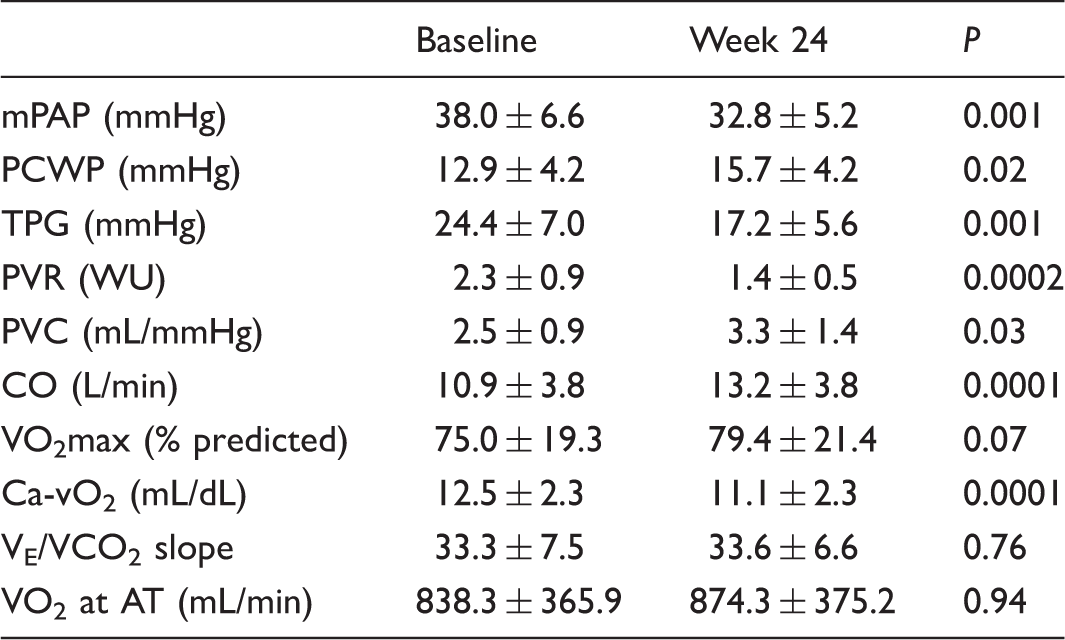
mPAP, mean pulmonary artery pressure; PCWP, pulmonary capillary wedge pressure; TPG, transpulmonary pressure gradient; PVR, pulmonary vascular resistance; PVC, pulmonary vascular compliance; CO, cardiac output; VO2max, maximum oxygen uptake; Ca-vO2, arteriovenous oxygen content difference; VE/VCO2, minute ventilation/carbon dioxide production; VO2 at AT, oxygen consumption at anaerobic threshold.
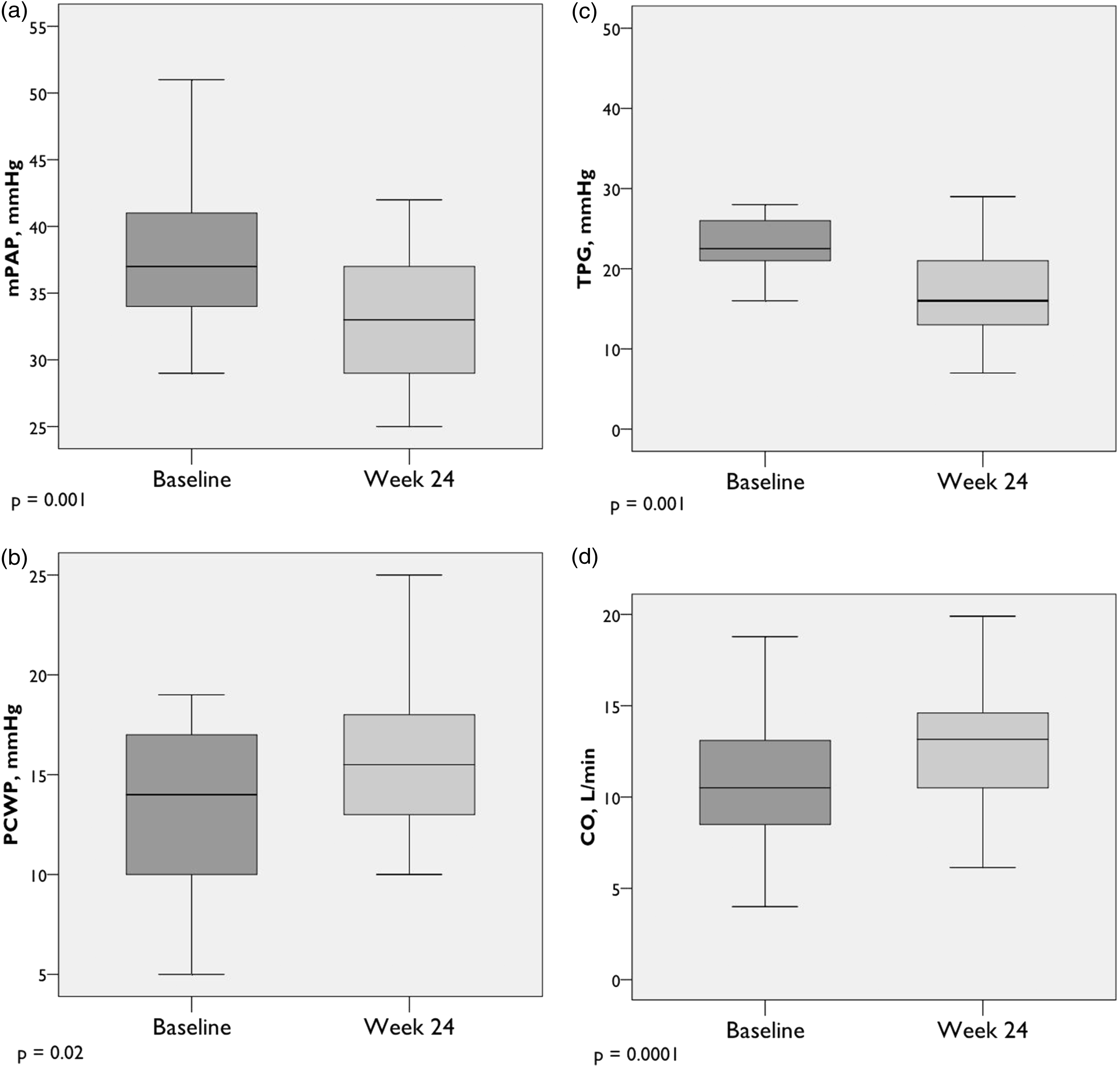
Peak exercise hemodynamics (n = 22). (a) Mean pulmonary artery pressure; (b) pulmonary capillary wedge pressure; (c) transpulmonary pressure gradient; (d) cardiac output. Box plots show median, IQR, minimum, and maximum values (± 1.5·IQR).
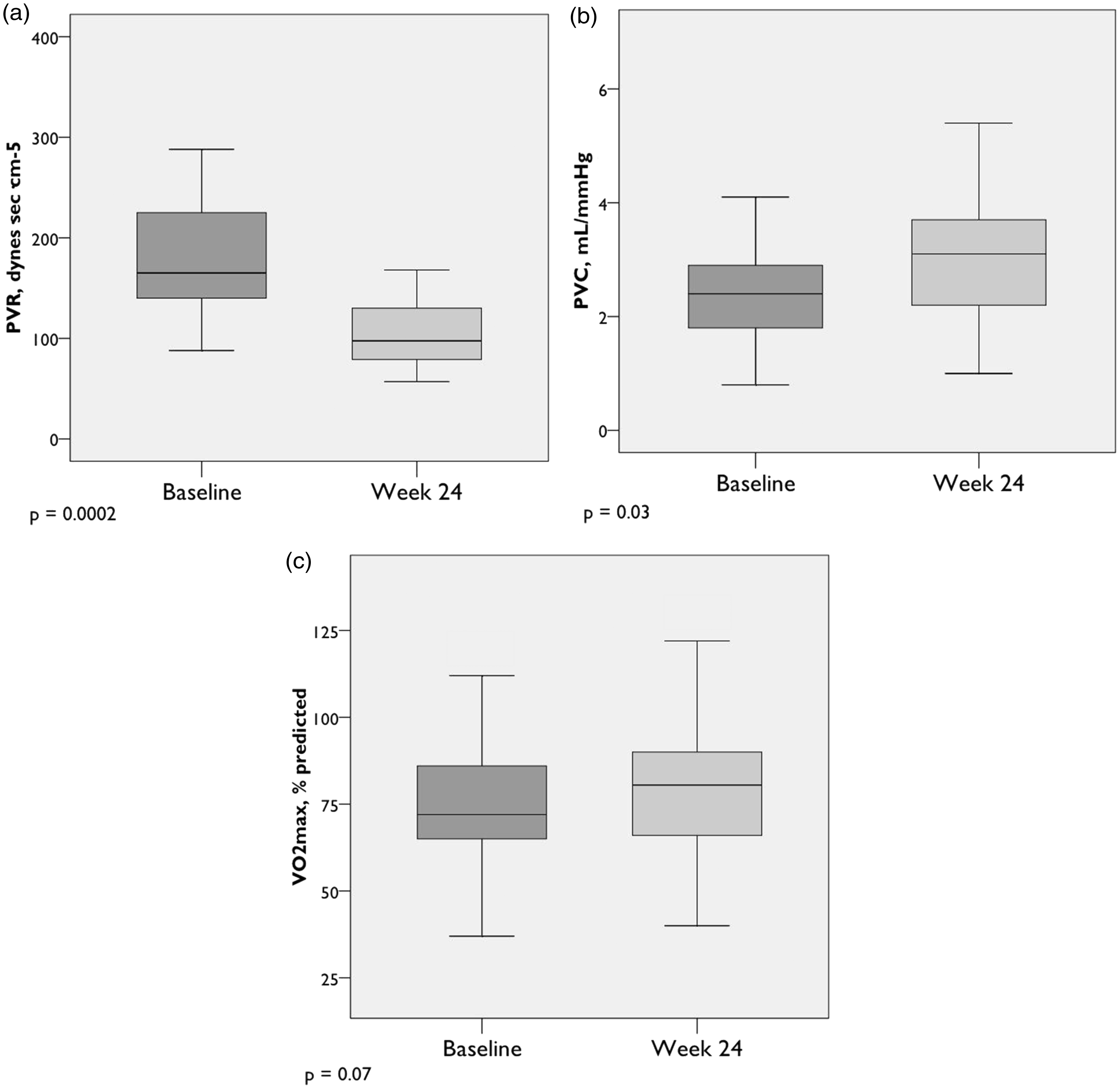
Peak exercise hemodynamics (cont.) (n = 22). (a) PVR = Pulmonary vascular resistance; (b) Pulmonary vascular compliance; (c) Maximum oxygen uptake. Box plots show median, IQR, minimum, and maximum values (±1.5·IQR).
Among the secondary endpoints, 6MWD improved by 34.8 ± 73.2 m (P = 0.04) after 24 weeks of treatment. In addition, perceived dyspnea during the six-minute walk test (6MWT) improved, from a baseline Borg Dyspnea Score of 5.1 ± 2.8 (intense breathlessness) to 3.0 ± 0.9 (moderate breathlessness) after 24 weeks (P = 0.002). A significant decrease in WHO FC was also observed, from 2.0 ± 0.6 at baseline to 1.5 ± 0.6 at study end (P = 0.002). Improvements in exercise capacity and FC were noticed by every patient at month 3.
Adverse events.*

Total number of events; in some participants who completed study >1 event occurred.
Participants withdrawn from study.
Eight participants failed to improve during treatment with ambrisentan and experienced subjective lack of improvement or worsening dyspnea on exertion. These participants were subsequently withdrawn from the study, between the week 4 and week 8 visits. Baseline characteristics of participants who were withdrawn from study, including resting and peak exercise hemodynamics, are presented in Table 2. End-of-study measurements of exercise hemodynamics were not assessed in these patients. Mean 6MWD of the last 6MWT carried forward was not significantly different from baseline (−21.1 ± 72.0 mmHg, P = 0.4). Five of the eight participants withdrawn were transitioned to a PDE-5 inhibitor for treatment of ePH. All five noted subjective improvement of their dyspnea on exertion, and demonstrated improved exercise capacity based on either 6MWD or submaximum exercise testing. Three of the eight participants who were withdrawn from the study were lost to follow-up.
Discussion
Our results suggest that treatment of ePH with ambrisentan is well tolerated among most and is associated with significant improvement of exercise pulmonary hemodynamics, symptoms, and functional status. Few studies have investigated the treatment of ePH patients. An open-label pilot study of the use of ambrisentan in 12 patients with ePH associated with systemic sclerosis (SSc) demonstrated an improvement in exercise hemodynamics and 6MWD over 24 weeks, with significant changes in exercise PVR, mPAP, and CO. 19 Park et al. found a significant increase in 6MWD among a small cohort ten patients after short-term (median 189 days) and long-term (median 416 days) treatment with bosentan or sildenafil. 20 Two additional pilot studies among patients with SSc-associated ePH showed improvements in hemodynamic parameters after treatment with bosentan.21,22 Ours is the first study to demonstrate the extended benefit of treatment of ePH with an ERA marked by an improved pulmonary vascular response to exercise.
In PAH, intimal proliferation and fibrosis, medial hypertrophy, and in situ thrombosis characterize the pathological findings in the pulmonary vasculature. 27 At earlier stages, vascular remodeling may be confined to the small pulmonary arteries, resulting in decreased compliance of the pulmonary vasculature. As pathologic remodeling progresses, PVR and right ventricular work increase, along with a corresponding increase in mPAP. Among patients with ePH, our study showed significant improvements of mPAP, PVR and PVC, and 6MWD and dyspnea after 24 weeks of treatment with ambrisentan, which may reflect prevention of progression of resting PAH.
Pulmonary vascular compliance, calculated as stroke volume over pulmonary arterial pulse pressure (SV/PP), is an accepted estimate of the total arterial compliance of the pulmonary vascular tree and reflects the ability of the vascular bed to distend in response to RV contraction and recoil during diastole. 28 Unlike PVR, which quantifies flow and resistance in the distal pulmonary arterial vascular bed, PVC reflects the elastic properties that modulate the impact of pulsatile blood flow. Decreased pulmonary arterial compliance and distensibility may represent early dysfunction and remodeling of the pulmonary vascular bed. 29 Previously, we found that PVC was reduced at rest, peak exercise and throughout recovery in ePH, 10 indicating a more pronounced and sustained pulmonary vascular stiffness compared to normal controls and those with exercise HFpEF. In the present study, a significant decrease in resistance was accompanied by a significant increase in compliance after six months of treatment. Interestingly, in 11 patients we evaluated PVC during recovery from peak exercise and found that the compliance recovery pattern had normalized. Taken together, these findings, at peak exercise and during recovery, could be attributed to vasodilation, or could suggest reverse remodeling of the distal pulmonary vascular bed in patients with ePH.
Patients with hemodynamic evidence of left ventricular diastolic dysfunction were excluded from the study and peak PCWP values were within the normal range (< 20 mmHg) at baseline and study end. Nevertheless, we observed a significant increase in peak PCWP after 24 weeks compared to baseline, which may reflect a positive effect of increased flow through the pulmonary circuit.
Peak VO2 is an important prognostic marker in PAH and defines exercise limitations that result from abnormalities in the cardiopulmonary circuit. In patients with PAH, VO2 is generally reduced and reflects the decreased ability to augment pulmonary and systemic blood flow during exercise. 27 In addition, as PVR increases, there is a decrease in CO and impaired oxygen delivery to skeletal muscle. Our results show significant improvements in CO consistent with improved flow through the pulmonary circuit and increased peak VO2.
Systemic oxygen extraction during exercise, reflected by a widening difference in Ca-vO2, normally increases threefold during short-term incremental exercise. Our results show a significant decline in Ca-vO2 at maximum exercise after ambrisentan treatment. This is currently an active area of investigation in our laboratory.
We also observed improvements among secondary endpoints, including WHO FC, 6MWD, and perceived breathlessness. At baseline, the majority of participants presented with FC 2, indicating a slight limitation of physical activity resulting in undue dyspnea or fatigue. All participants who completed study improved in functional class after six months of treatment, able to perform ordinary physical activity without limitation. Exercise capacity, as assessed by 6MWD, also improved significantly after the treatment period. On average, participants completed the 6MWT with only moderate dyspnea at study end, compared to intense dyspnea at baseline. Notably, it took an average of three months for participants to notice a subjective improvement in their exercise capacity and dyspnea on exertion. This observation builds on our hypothesis that treatment of ePH with ambrisentan results in ongoing reverse remodeling of the pulmonary vascular network, which needs to reach a certain threshold before a difference in functional status is noticed.
Limitations
While results are promising, important limitations of the present study must be considered. This is a single-center, open-label trial where all patients were referred to our pulmonary vascular disease program based on the results of a prior clinically indicated iCPET. Results should be interpreted with caution in view of the small sample size and the lack of a control group. However, reports such as this can provide valuable information about current clinical practices and can guide the design of randomized controlled trials. End-of-study data were not collected from the eight participants withdrawn due to subjective lack of improvement and subjective worsening dyspnea on exertion. However, those patients transitioned to a PDE-5 inhibitor then realized improvement, which supports our hypothesis that patients with ePH represent an early and potentially readily treatable disease state.
Open label designs also raise concern for the introduction of bias in the results as it is difficult to control for benefits from study enrollment that are unrelated to study treatment, such as more frequent follow-up and increased disease awareness. While bias may have influenced secondary endpoints, it is very unlikely to have had any effect on invasively measured cardiopulmonary hemodynamics. A single-center, single-operator study design allows for an accurate comparison of data from baseline to study end and minimizes the potential for performance bias. While the need for a larger randomized controlled trial is justified, the prevalence of ePH and ability to perform iCPETs at other institutions pose considerable difficulties.
Of note, a recently published study has proposed a revised definition of ePH which is based on age-specific upper limits of normal for maximum exercise pulmonary hemodynamics. 9 In the current study, we chose to continue participant recruitment under original inclusion criteria which included a lower upper limit of normal for PVR max (<1.0 WU), for enrollment consistency. Only two of 22 participants included in the analysis of this study had a low baseline PVRmax, which would not affect overall trends and significant changes reported.
Conclusion
Patients with ePH may provide a unique window into the pathogenesis of PAH, as an early phase of disease with an abnormal pulmonary vascular response to exercise. Our findings suggest that treatment of ePH with ambrisentan results in improved cardiopulmonary hemodynamics and functional status over a six-month period, on average. Treatment of ePH may prevent the progression of vascular remodeling and development of established PAH. Further study of ePH treatment is warranted, especially with regards to disease progression, functional capacity, and quality of life.
Footnotes
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
Gilead Sciences, Inc. provided funding for the study. Gilead Sciences had no involvement in the design and conduct of the study or the interpretation of the results.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.