Rare Tumors is an international peer-reviewed medical journal established in 2009. The journal is focused on rare cancers and aims to expand upon current knowledge on their presentation, diagnosis, management, and outcomes. We reviewed the 335 case reports published from 2009 to 2015. We found great diversity in both the country of origin as well as specialty of first authors. Outside of the United States (US) and European Union (EU), there were 20 countries with contributions to the journal. Similarly, there was representation from twelve medical specialties with first authorship of reports. Rare Tumors continues to encourage involvement from physicians across the globe and from all medical disciplines.
Rare Tumors is an international, open access peer-reviewed medical journal with a primary focus on the study of rare neoplasms. It was established in 2009 in Pavia, Italy. Rare malignancies are by definition, rare, and this complicates their study through traditional mechanisms such as clinical trials. The journal aims to expand upon current knowledge on the presentation, diagnosis, management, and outcomes of uncommon tumors that cannot be studied by conventional means. It provides a venue for authors to publish their experience in the treatment of rare cancers in order to improve the care of future patients. As an online, open access publication, the journal functions as a global, public repository for experience in the modern treatment of rare tumors that is available to anyone with an interest in their study and treatment.
To better characterize the nature of this diverse group of tumors, we analyzed publication patterns in Rare Tumors.
Materials and Methods
We retrospectively reviewed all the published reports in Rare Tumors, dating from 2009 to 2015, including volumes one through seven. We analyzed each of the published case reports, examining the key points of interest that would enable us to evaluate statistical trends and observe the diversity of published work. Pertinent data on the patients’ cases as well as the authors were abstracted and evaluated. Specifically, we reviewed patient demographics, tumor characteristics, and treatment modalities. Additionally, we recorded information on country of origin, number and specialty of authors, and date of publication. Data was collected from each publication and the final database was reviewed by the authors. The mean or most prevalent results of each category were calculated and documented.
Results
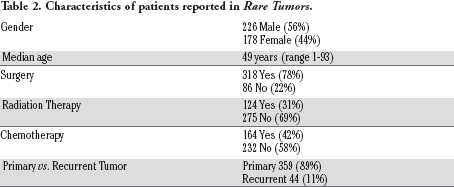
There have been a total of 335 case reports and case series of 404 patients published in Rare Tumors at the time of our analysis in July of 2015. Tumor histology is listed in Table 1. Histologically, no single tumor type predominated. Carcinoma, not otherwise specified, and adenocarcinoma, made up 27% of all cases. This was followed in frequency by sarcomas in 9.7% and lymphoma in 6.7% of cases. The remaining two-thirds of cases were divided into 39 distinct histologies. The primary tumor site anatomically is listed in Figure 1. Patient characteristics are listed in Table 2. Male patients were in a slight majority. The average age was low at 47 years. Surgery was utilized in the majority of cases and chemotherapy and radiotherapy in the minority. A total of 127 different chemotherapy regimens were reported, with no single regimen reported in more than 5 cases. The five most common drugs in decreasing order of frequency are cisplatin, etoposide, carboplatin, doxorubicin, and gemcitabine. A world cloud in Figure 2 depicts the relative frequency of the usage of each individual agent. Approximately 25% of cases reported the clinical course of the patient until death. Median follow-up for all cases was 14 months (range 1 to 264 months). Seventy-five percent of cases had at least 6 months of follow-up and 25% of cases had at least 36 months of follow-up. Approximately 11% of patients had a documented recurrence of their tumors. Median time to recurrence was 12 months (range 0 to 264 months). The fragmented nature of the data by histology, site, and treatment modality prevented a statistical analysis of risk factors for recurrence and death from malignant progression.
Histological characteristics of tumor types reported in Rare Tumors.
Histological type
N.
%
Carcinoma, not otherwise specified
55
16.7
Adenocarcinoma
34
10.3
Sarcoma
32
9.7
Lymphoma
22
6.7
Adenoma
18
5.5
Blastoma
12
3.6
Squamous cell carcinoma
10
3.0
Cytoma
9
2.7
Angioma
8
2.4
Mesothelioma
8
2.4
Small Cell Carcinoma
8
2.4
Angiosarcoma
7
2.1
Fibroma
6
1.8
Giant Cell Tumor
6
1.8
Melanoma
6
1.8
Neuroendocrine carcinoma
6
1.8
Schwannoma
6
1.8
Germ cell tumor
5
1.5
Langerhans cell histiocytosis
5
1.5
Lipoma
5
1.5
Angiomyxoma
4
1.2
Granuloma
4
1.2
Hemangioendothelioma
4
1.2
Liposarcoma
4
1.2
Renal Cell Carcinoma
4
1.2
Carcinoid
3
0.9
Dendritic cell carcinoma
3
0.9
Ewing's sarcoma
3
0.9
Fibrous tumor
3
0.9
Meningioma
3
0.9
Osteosarcoma
3
0.9
Rhabdoid
3
0.9
Desmoid
2
0.6
Ependymoma
2
0.6
Epithelioid cell tumors
2
0.6
Hyperplasia
2
0.6
Kaposi's sarcoma
2
0.6
Merkel cell carcinoma
2
0.6
Parachordoma
2
0.6
Rhabdomyosarcoma
2
0.6
Thymoma
2
0.6
Fibroelastoma
1
0.3
Glioma
1
0.3
Frequency of tumor site by primary organ system.
Characteristics of patients reported in Rare Tumors.
Gender
226 Male (56%)
178 Female (44%)
Median age
49 years (range 1-93)
Surgery
318 Yes (78%)
86 No (22%)
Radiation Therapy
124 Yes (31%)
275 No (69%)
Chemotherapy
164 Yes (42%)
232 No (58%)
Primary vs. Recurrent Tumor
Primary 359 (89%)
Recurrent 44 (11%)
Word cloud representation of frequency of chemotherapy usage.
The medical specialty of first author of each report is represented in Figure 3. Their country or region of origin is shown in Figure 4. Most publications were from the United States with 113 (34%) cases, European Union with 76 (22.4%) cases, and Japan with 43 (13%) cases.
Frequency of authors by medical specialty.
Frequency of authors by country of origin.
Case Reports
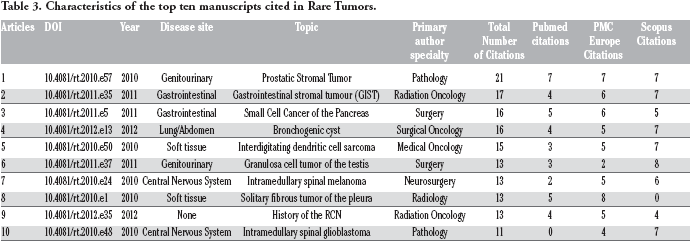
The ten most cited papers are summarized in Table 3. Nine are case reports or case series and one is a summary of the history of the Rare Cancer Network. No single theme predominates in the type of tumor, anatomic location, or specialty submitting the case reports that rank in the ten most cited papers. The one commonality present is that established evidence based guidelines do not exist to guide the treatment of the malignancies presented in nine of ten cases. The case report of a gastrointestinal stromal tumor (GIST) is an exception to this. For a very rare tumor type such as small cell carcinoma of the pancreas, there is little prospective research in the medical literature to guide practitioners. The five most cited papers are summarized below.
Characteristics of the top ten manuscripts cited in Rare Tumors.
Articles
DOI
Year
Disease site
Topic
Primary author specialty
Total Number of Citations
Pubmed citations
PMC Europe Citations
Scopus Citations
1
10.4081/rt.2010.e57
2010
Genitourinary
Prostatic Stromal Tumor
Pathology
21
7
7
7
2
10.4081/rt.2011.e35
2011
Gastrointestinal
Gastrointestinal stromal tumour (GIST)
Radiation Oncology
17
4
6
7
3
10.4081/rt.2011.e5
2011
Gastrointestinal
Small Cell Cancer of the Pancreas
Surgery
16
5
6
5
4
10.4081/rt.2012.e13
2012
Lung/Abdomen
Bronchogenic cyst
Surgical Oncology
16
4
5
7
5
10.4081/rt.2010.e50
2010
Soft tissue
Interdigitating dendritic cell sarcoma
Medical Oncology
15
3
5
7
6
10.4081/rt.2011.e37
2011
Genitourinary
Granulosa cell tumor of the testis
Surgery
13
3
2
8
7
10.4081/rt.2010.e24
2010
Central Nervous System
Intramedullary spinal melanoma
Neurosurgery
13
2
5
6
8
10.4081/rt.2010.e1
2010
Soft tissue
Solitary fibrous tumor of the pleura
Radiology
13
5
8
0
9
10.4081/rt.2012.e35
2012
None
History of the RCN
Radiation Oncology
13
4
5
4
10
10.4081/rt.2010.e48
2010
Central Nervous System
Intramedullary spinal glioblastoma
Pathology
11
0
4
7
Colombo et al. present an unfortunate case of prostatic stromal tumor in a 34-year-old male. These tumors arise from specialized hormone-dependent mesenchymal cells. This patient was diagnosed with a stromal tumor with unknown malignant potential (STUMP) after presenting with lower urinary tract symptoms and undergoing a transurethral resection. The tumor cells are immunoreactive for vimentin, progesterone receptor, and CD34. He subsequently underwent a radical prostatectomy followed by radiotherapy receiving 60 Gray (Gy) in 30 fractions delivered to the prostate bed. Unfortunately, the patient developed lung metastases. These lung lesions were responsive to ifosfamide and liposomal doxorubicin, but the patient ultimately progressed and expired 25 months after the initial diagnosis.1
Knowlton and colleagues discuss a patient with unresectable GIST. In the United States, 4500 to 6000 cases of GIST are diagnosed each year. Due to the unresectable nature of this disease, the reported patient underwent a debulking and remained symptom-free for four years. He then had local progression for which he underwent palliative radiation therapy receiving 36 Gy in 24 fractions. At 20 years follow-up, the patient was disease free from GIST, but he did develop rectal adenocarcinoma. Although an outlier in terms of success of therapy in comparison to the majority of patients with recurrent GIST after surgery, the case highlights extended survivals is possible for some patients.2 Six cases of resectable pancreatic small cell carcinoma are detailed in small case series by Winter et al. There are only 30 cases of small cell pancreatic cancer reported to date in literature. The six patients in this report had tumors in the head of the pancreas and all underwent pancreaticoduodenectomy. All six patients received adjuvant therapy including radiation therapy and/or chemotherapy with cisplatin and etoposide. The authors found a median survival of 20 months (9-173 months), which was higher than prior reports with a median survival of six months with surgical resection alone.3 Goevarts and colleagues report a case of a bronchogenic cyst presenting as an unusual retroperitoneal cystic mass. The authors noted that only 30 cases of retroperitoneal bronchogenic cysts have been documented in the literature to date when strict diagnostic criteria were used to screen reports in the literature.4 Weiss and colleagues report an unusual case of interdigitating dendritic cell sarcoma (IDCS) in the axilla. A 25-year-old female presented with an enlarged right axillary lymph node. The patient underwent an excisional biopsy. The tumor stained positive for S100 and smooth muscle actin and negative for MelanA. Flow cytometry was done and showed no monoclonality and ruled out leukemia or lymphoma. Imaging with Positron Emission Tomography/Computed Tomography (PET/CT) showed low-level uptake in the axilla in the area of excision but no other abnormalities. An independent pathologic review led to a diagnosis of IDCS. The patient has been followed with PET/CT scans with no evidence of disease recurrence at 24 months.5
Discussion and Conclusions
Rare Tumors is an open access, online medical journal and reference site for rare cancers. This review provides a look into the case reports published to date. Prior to the year 2000, there were few resources available for patients and physicians dealing with rare forms of cancer in comparison to those available for more common tumors types. However, the development of the Internet has provided a variety of ways for research networks and patient advocacy organizations to create collaborative mechanisms on a scale impossible before the advent of digital collaboration. Rare Cancer Network was established in 1993 to provide a framework for institutions across the globe to work together on rare cancer research with a specific focus on the role of radiation therapy. The network consists of 130 investigators in 24 countries.6 The International Rare Cancer Initiative was formed through a collaboration of the European Organisation for the Research and Treatment of Cancer (EORTC), Cancer Research United Kingdom (UK), the National Institute for Health Research Cancer Research Network (NCRN), and the United States National Cancer Institute (NCI). This organization aims to develop clinical trials of treatments for rare cancers.7 In the United States, the National Organization of Rare Disorders (NORD) is a patient advocacy organization committed to staying current on the identification and treatment of rare disorders.8 In the European Union, the European Organization for Rare Diseases plays a similar role.9 The US National Institute of Health (NIH) also has a rare disease sector that targets patients with rare diseases, aiding research and allocating funds to study rare diseases.
In our analysis, we have found a wide variety of cases presented by a diverse group of physicians. The top three nations contributing to the global scientific literature, by frequency of publication, are the US, China and UK.10 In comparison, the top three origins of case reports in Rare Tumors are, in descending order, USA, EU and Japan. Outside of the United States and European Union, there were 20 countries with contributions to the journal. Most publications represented a single geographic site, with some significant exceptions of multi-national collaboration in publication.11,12 Additionally, open access publishing represents a mechanism by which developing world authors can play an important role in contributing to the development of the literature of medical science.
As expected, the average patient reported in Rare Tumors differed from the typical patient with a malignancy in the United State and Western European. The typical patient age was younger than that of common malignancies, 49 years of age in Rare Tumors versus 67 years of age for all tumor types in the United States.13 The top five primary tumor sites in Rare Tumors were gastrointestinal, genitourinary, soft-tissue/bone, gynecology, and head and neck tumors. The top five tumor sites for all malignancies in the USA in 2014 were Genitourinary, Gastrointestinal, Respiratory, Breast, and Lymphatic system malignancies.14
While case reports do not represent a high rank of scientific evidence, for many rare diseases it is the only literature available for physicians to use in guiding their decision-making. When evaluating such case reports, it is critical to remember their limitations, particularly in that unusual presentations or unexpected outcomes may prompt an investigator to publish their report in greater proportion than similar cases without exceptional outcomes. This can lead to both an overestimate of the benefits of interventions as well as an incorrect estimate of the risks of therapy when exceptional toxicities or disease progression are reported. However, it is important to continue publishing these reports to further detail and share our understanding and experiences of these uncommon cancers. We encourage involvement from physicians around the world and from all medical disciplines.
Footnotes
Acknowledgements
Professor Camillo Porta of Pavia, Italy for the original conception of the journal.
References
1.
ColomboPCeresoliGLBoiocchiL. Prostatic stromal tumor with fatal outcome in a young man: histopathological and immunohistochemical case presentation. Rare Tumors2010;2:e57.
2.
KnowltonCABradyLWHeintzelmanRC.Radiotherapy in the treatment of gastrointestinal stromal tumor. Rare Tumors2011;3:e35.
3.
WinterJMNarangAKMansfieldAS. Resectable pancreatic small cell carcinoma. Rare Tumors2011;3:e5.
4.
GovaertsKVan EykenPVerswijvelGVan der SpeetenK.A bronchogenic cyst, presenting as a retroperitoneal cystic mass. Rare Tumors2012;4:e13.
5.
WeissGJAlarconAHalepotaM. Molecular characterization of interdigitating dendritic cell sarcoma. Rare Tumors2010;2:e50.
6.
PatelAOzsahinMMirimanoffRO. The rare cancer network: achievements from 1993 to 2012. Rare Tumors2012;4:e35.
7.
International Rare Cancer Initiative. Introduction. 2015[cited 2015 12 August]; Available from: http://www.irci.info/.
Europen Organisation for Rare Diseases. [12 August 2015]; Available from: www.eurordis.org.
10.
WareM.The STM Report. Netherlands: International Association of Scientific, Technical and Medical Publishers Prins Willem Alexanderhof 5, The Hague, 2595BE; 2015.
11.
MirimanoffR-OOzsahinMThariatJ. History of the rare cancer network and past research. Rare Tumors2014;6:5462.
12.
OzsahinMMirimanoffR-OThariatJ. The rare cancer network: ongoing studies and future strategy. Rare Tumors2014;6:5465.
13.
National Cancer Institute. Median age of cancer patients at diagnosis, 2000-2003. Surveillance, epidemiology, and end results program (SEER); 2000–2003.
14.
SiegelRLMillerKDJemalA.Cancer statistics, 2015. CA Cancer J Clin2015;65:5–29.