
Abstract
Gliosarcoma (GS) is a rare and exceedingly malignant neoplasm of the central nervous system. It displays clinical features similar to glioblastoma, yet is histologically unique as it harbors both gliomatous and sarcomatous cellular components. Involvement of the neuro-axis is predominantly limited to the cerebral parenchyma and meninges. Primary GS of the spinal cord is rarely encountered. We report a case of a 54 year old male who presented with 2 months of progressive, bilateral lower extremity sensory deficits. Magnetic resonance imaging of the neuro-axis revealed multiple intradural lesions involving the cervical and thoracic spinal cord without evidence of intracranial involvement. Surgical resection of a dural based, extramedullary cervical lesion and two exophytic, intramedullary thoracic lesions revealed gliosarcoma, WHO grade IV. The patient died approximately 11 months after presentation. This report confirms that GS is not limited to supratentorial involvement and can primarily affect the spinal cord.
Introduction
Gliosarcoma (GS) is an exceedingly rare and malignant neoplasm of the central nervous system most commonly affecting the cerebrum. Primary GS of the spinal cord has been reported only once. 1 Here we report a case of primary, multifocal spinal GS without intracranial involvement.
Case Report
Presentation
A 54 year old, other-wise healthy male presented with 2 months of progressive lower extremity sensory dysfunction and gait instability. Physical examination revealed decreased sensation to pinprick along the anterior aspect of the right thigh as well as decreased proprioceptive sense at the great toe. Neurological examination, including motor function and reflexes, was otherwise normal.
Initial Work-Up
Magnetic resonance imaging (MRI) of the entire neuro-axis revealed numerous contrast enhancing, intradural, extramedullary lesions involving the cervical and thoracic spinal cord (Figure 1) as well as a single lesion adjacent to the conus medullaris (Figure 2). Many of these lesions caused compression and associated edema of the adjacent spinal cord. Subsequent MRI of the brain with and without gadolinium infusion failed to show any evidence of intracranial involvement. Computed tomography of the chest, abdomen and pelvis with intravenous contrast was unrevealing. Cerebrospinal fluid (CSF) obtained via lumbar puncture revealed a protein level of 300 mg/dL and a relative lymphocytosis. Flow cytometry was negative for monoclonal proliferation or other malignancy.
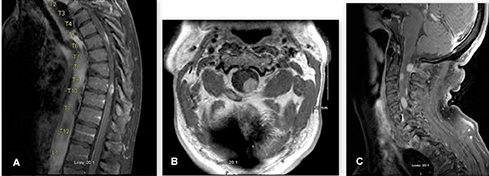
Sagittal thoracic (A), axial cervical (B) and saggital cervical (C) planes of T1 weighted magnetic resonance images showing multiple contrast enhancing, intradural, extra-medullary lesions.

Sagittal T1 weight magnetic resonance images of the lumbar spine showing a single, contrast-enhancing lesion at the level of the L1 vertebral body.
Intervention
An open biopsy of the C1 lesion was performed as this lesion appeared to be the most accessible. Intra-operatively, the lesion was adherent to the dorsal dura, but cleanly separated from the spinal cord. The patient tolerated the procedure well with no immediate post-operative changes. Three days post-operatively, the patient's neurological function abruptly declined with the development of lower extremity weakness and profound sensory dysfunction from the T8 level caudally. Gait became significantly unstable. Pathologic diagnosis was not confirmed at this time and the patient elected to undergo exploration and decompression at the level of his mid thoracic cord as his symptoms were referable to these lesions. He returned to the operating room for T6-T9 laminectomy and resection of intradural T6 and T9 lesions. These lesions were found to be exophytic from the spinal cord parenchyma without a clear tissue plane of separation. The exophytic portions of the lesions were resected. The patient's neurologic function was unchanged immediately post-operatively, but steadily declined to the point of complete paraplegia over the course of one month.
Pathology
Surgical specimens from both resections were identical and demonstrated a high grade neoplasm with intimate interdigitation (marmoreal pattern) of cytologically-malignant fibrillary to epithelioid glial cells and spindled sarcomatous cells on hematoxylin and eosin (Figure 3A) and reticulin stain (Figure 3B). The glial component was strongly immunoreactive for glial fibrillary acidic protein (GFAP) (Figure 3C) and S100, and negative for IDH-1 and epithelial membrane antigen, while the reticulin-rich sarcomatous areas were negative for smooth muscle actin and desmin, with only small numbers of entrapped/admixed GFAP and S100 positive glial cells. Profuse microvascular proliferation was present and both components showed elevated MIB-1 cell cycle labeling of >10-12% (Figure 3D). Gliosarcoma, WHO grade IV, was diagnosed. Fluorescence in situ hybridization studies demonstrated positive loss of PTEN and monosomy 10, with a single cell manifesting amplification of EGFR, although areas showed high polysomy of chromosome 7 sequences (>5 copies, 30% of cells). Electron microscopy showed neoplastic cells with broad cytoplasmic processes containing large bundles of intermediate filaments; no intercellular junctions or other features of meningothelial cell differentiation were observed.

A) Photomicrograph showing the two components in the gliosarcoma: cytologically atypical nests of glial cells (arrows) are intimately admixed with spindled sarcomatous tumor (Hematoxylin and Eosin, 200×). B) Reticulin stain better differentiates the reticulin poor nests of malignant glial cells (arrows) from the reticulin-rich sarcomatous component (modified Gordon and Sweets reticulum II stain [Ventana Corp, Tuscon, AZ], 200×). C) Immunostaining for GFAP shows strong immunoreactivity in the malignant glial component (arrows), with only occasional entrapped immunoreactive cells within the spindled sarcomatous component (Immunostaining for GFAP [Dako Corp., Carpinteria, CA], with light hematoxylin counterstain, 200×). D) Elevated MIB-1 cell cycle labeling in both the malignant glial and sarcomatous (arrow) components (200×).
Post-Operative Course
Post-operatively, the patient received adjuvant radiation therapy to the cervical, thoracic and lumbar spine with concurrent Temozolomide treatment. Despite this, he developed worsening leptomeningeal disease and progressive weakness of the upper extremities. He was then trialed on avastin therapy and failed to show a significant clinical response. He died approximately 11 months after his initial presentation.
Discussion
Gliosarcoma is an exceedingly rare primary central nervous system tumor. Since it was first reported by Stroebe in 1895, there have only been a handful of retrospective case series describing the natural history of GS. Histologically, GS shares many features with glioblastoma (GBM), however, immunohistochemical analysis shows a biphasic architecture with an astrocytic component staining positive for GFAP and a sarcomatous portion, commonly composed of spindle cells, which stains positive for reticulin and negative for GFAP. GS accounts for an estimated 2.2% of all malignant gliomas. 2 Treatment is identical to that of GBM, generally involving surgical resection with concurrent radiation and temozolomide therapy. Prognosis is bleak with a mean survival of 9 months and, like GBM, is affected by age of presentation, extent of surgical resection and adjuvant radiation therapy. 2 GS is found almost exclusively in the supratentorial space, with a predilection for the temporal lobes. 2 Nonetheless, variation in the anatomical location of GS has been observed in both cases of metastatic disease as well as uncommon primary presentations. Metastasis of GS to the spinal cord is rare, but established in the literature. 3 Primary spinal GS, in contrast, is not well described. An extensive literature search revealed only one previous case of primary spinal GS reported by Barbagallo et al. 1 Surgical resection in that case revealed an extramedullary, intradural lesion densely adherent to the meninges, yet with no involvement of the spinal cord parenchyma.
Our patient presented with a primary, multifocal GS of the spinal cord and leptomeninges. Both multi-focal presentation and meningeal involvement have been reported in the literature. 4 In fact, Han et al. found that those patients with GS mimicking meningiomas were found to have better outcomes, likely due to a higher rate of gross total resection in that patient group. 5
Unlike the case reported by Barbagallo et al., the GS described here was found to be intimately involved with the spinal cord parenchyma and to be multi-focal in distribution. We hypothesize that the initial lesion likely originated from the spinal cord parenchyma and secondarily spread to the meninges, causing diffuse leptomeningeal disease with numerous foci of adherence to the adjacent spinal cord.
Conclusions
Primary GS is a rare, but malignant neoplasm of the central nervous system. Even rarer, is primary involvement of the spinal cord. To the best of our knowledge, this is the first case of primary, multi-focal spinal GS described in the literature. More importantly, this report confirms that GS is not limited to supratentorial involvement and should be included within the differential diagnoses for the work-up of primary spinal cord lesions.
Footnotes
Acknowledgements
The authors thank Ms. Lisa Litzenberger for photographic expertise and Dr. Bette Demasters for her assistance in the diagnosis of this case.