
Abstract
Juvenile polyposis syndrome (JPS) is an infrequent autosomal dominant hereditary predisposition to the occurrence of hamartomatous polyps in the colon and rectum. We describe the case of a 12-year-old boy with JPS associated with an abdominal tumor. Histological sections of the abdominal tumor showed components of adenocarcinoma, osteosarcoma, and choriocarcinoma. Immunohistochemistry was AE1/AE3, CK7, HCG and SALL4 positive. Juvenile polyposis syndrome patients are at increased risk of colorectal adenocarcinoma. However, we present a case of an adenocarcinoma associated with other unusual components. This association has not been reported before.
Introduction
Juvenile polyposis syndrome (JPS) is a rare dominant autosomal hereditary disorder characterized by the presence of five or more juvenile polyps along the colorectum; or juvenile polyps throughout the gastrointestinal tract or any number of polyps in patients with family history of juvenile polyposis. 1 Carcinosarcoma is a mixed biphasic tumor, characterized by a carcinomatous component and a sarcomatous component, an extremely rare and aggressive entity with poor prognosis. 2 Carcinosarcoma has been described in the gastrointestinal tract predominantly in the esophagus, in the stomach and in the biliary tract, whereas the presence of this type of tumor in the colon has been reported only rarely. 3 We report the case of a 12-year-old boy with JPS associated to a colon tumor characterized as a conventional adenocarcinoma exhibiting areas of choriocarcinomatous and osteosarcomatous elements.
Case Report
A 12-year-old boy who presented with an abdominal mass associated with B symptoms as well as anemia, requiring several blood transfusions, and no family history of gastrointestinal problems. On initial examination a large palpable ill-defined mass, fixed to deep underlying structures was found in the right flank. The mass was characterized by an abdominal computed tomography (CT) scan (Figure 1) that revealed a heterogeneous stenotic mass affecting the cecal lumen and compromising the colon. Severe hepatomegalia was observed including several small nodular lesions corresponding to metastasis.
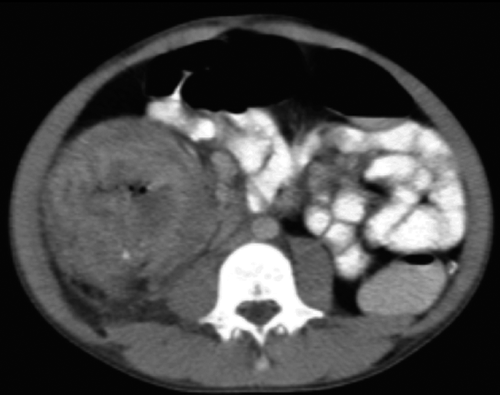
Marked thickening of the ascending colon wall affecting 90% of the lumen with the resulting luminal stenosis.
The patient underwent a right hemicolectomy. A 10×8 cm stenotic mass with 100% luminal obstruction at the ileocecal valve was identified along with polypoid lesions compatible with juvenile polyposis (Figure 2A). The mass had areas consistent with a conventional malignant adenocarcinoma (Figure 2B) with large areas of choriocarcinomatous differentiation (60%) (Figure 2C) and osteosarcomatous differentiation (40%) (Figure 2D) as well as, evidence of vascular invasion, and no evidence of nodal spread (32 lymph nodes).

Hematoxylin and Eosin 400× shows: A) Juvenile polyps with high and low-grade dysplastic changes. B) Components of conventional adenocarcinoma. C) Section of osteosarcoma component. D) Section of germinal tumor.
Immunohistochemistry was AE1/AE3, CK7, HCG and SALL4 positive (Figure 3), and HEPAR, AFP, OCT3/4, CK20, S100, PLAP, CD117, CDX2, AML, and CD99 negative.

Immunohistochemistry: A) reactivity to cytokeratin (AE1/AE3) in the adenocarcinoma component; 4×. B) Reactivity to CK7 in the adenocarcinoma component; 50×. Reactivity to HCG is positive in the germinal tumor component; 400×.
At the time the pathology report was received, the boy remained in critical condition and was discharged at the request of the family and due to his overall poor prognosis. Thus, a follow-up of this case was not possible and the surgical treatment outcome was not known.
Discussion
Juvenile polyposis syndrome is a rare autosomal dominant disorder. 1 The first histological description of JPS was published in 1939 in a 30-month old female patient with rectal polyps. 4 The incidence is 1 in 100.000 births and is characterized by the presence of five or more juvenile polyps along the colon and rectum; or juvenile polyps throughout the gastrointestinal tract or any number of polyps in patients with family history of juvenile polyposis. 1 In the digestive tract the most frequent localization is the colorectum (98%), followed by the stomach (14%), the small intestine (7%) and less frequently in the duodenum (2%). 5 A retrospective study performed in a pediatric population described that the mean age at diagnosis was 4.7+7-2.5 years, predominantly in males with a ratio of 1.9:1. Symptoms include recurrent abdominal pain, protrusion of a polyp through the anus, diarrhea, and anemia. 6 Histological description of the lesions includes hyperplasia, dilated irregular glands, mucus filled cystic glands, edematous lamina propria with an inflammatory infiltrate, granulation tissue, absence of muscle fibers, and abundance of stroma. The diameter of the polyps ranges from 1 mm to a few centimeters. 7
A couple of mutations which interfere in the transforming growth factor beta (TGF-β) signal transduction in the germ cell have been suggested as potential causes for these lesions. Mutations in the SMAD4 and BMPR1A genes are among said mutations. These mutations are found in 50-80% of JPS cases.5,7 This syndrome, as well as other polyposis syndromes, poses as predisposing factors in the development of colorectal cancer in up to 39%. 8 Similarly, this syndrome has been reported to increase the risk for developing cancer in the pancreas, stomach and small intestine by 10-15%. The association between JPS and carcinosarcoma, choriocarcinoma and osteosarcoma has not been reported before.
Carcinosarcoma is an uncommon neoplasm comprising, carcinomatous and sarcomatous elements. It is also called sarcomatoid carcinoma, carcinoma with mesenchymal stroma, carcinoma with sarcomatous change, spindle cell carcinoma and pleomorphic anaplastic carcinoma. 9 This entity is rare in children; in fact, the first documented report was published in 2008. 10 It localizes most commonly in the head, neck, and female urogenital system. 3 In fact, only 23 cases of colon malignancies of this type have been reported in the literature. 3
In 2012, Ryu et al. reviewed the clinicopathological features of 17 reported cases of colonic sarcomatoid carcinomas. Mean age was 67.6 years. Ten patients were women. Some of these tumors, deeply invaded the bowel wall, metastasized to distant organs, resisted multi-agent chemotherapy and caused early patient death. Metastases were more frequent to the liver, lymph nodes, omentum, peritoneum and spleen. 11
The association of carcinosarcoma and choriocarcinoma has only been reported on the female urogenital system and bladder,12,13 making our case a clinicopathological challenge.
Hematoxylin & Eosin-based diagnosis may be difficult in these cases, which comprise various tumor components, thus, immunohistochemical diagnostic analyses can be a useful tool in an attempt to define the histogenesis of the tumor. In this case, we observed AE1/AE3 and CK7 positivity for epithelial components and SALL4 positivity for germinal components. Immunohistochemistry is extremely important for diagnosing these tumors. A choriocarcinoma was diagnosed based on the morphologic features and the presence of HCG and negativity of other germinal markers such as AFP, OCT3/4, PLAP, and CD117. 14
Conclusions
In summary, a case like the one presented here, of JPS associated with a conventional adenocarcinoma with choriocarcinomatous and osteosarcomatous differentiation has not been reported before. These changes may be explained by alterations in the TGF-β pathway. Genes involved in JPS such as SMAD4 and BMPR1A are also involved in the TGF-β pathway. TGF-β plays a crucial role in many cellular processes, including proliferation, differentiation, adhesion and migration. Disruptions in the TGF-β pathway have been reported in a uterine carcinosarcoma and choriocarcinoma.15,16 The role of TGF-β1 is associated with the initiation of the trophoblastic invasion process in choriocarcinoma by down regulation of the SMAD4 gene. 16 In addition, mutations in the SMAD4 gene have been associated with predisposition to germinal tumors and other tumors.17–19
Footnotes
Acknowledgments
The authors express their gratitude to Elizabeth A. Montgomery. They also would like to thank the Radiology Department at Hospital Infantil de San José, Bogotá, Colombia.