
Abstract
The dedifferentiated giant-cell tumor of the bone is a very rare variant of the giant-cell tumor (GCT). We report the clinical, radiographic and histological findings of a dedifferentiated GCT in which the dedifferentiated component consisted of small round cells. We also comment on previously reported cases of dedifferentiated GCT, discuss the clinical implications of this dual histology, and analyze the information published about the coexistence of similar genetic abnormalities in GCT and small round cell tumors of the bone.
Introduction
The giant-cell tumor of bone (GCT) is considered a benign, yet locally aggressive neoplasm. This tumor occurs in skeletally mature patients and affects women more often than men.1,2 The classical GCT of the bone usually affects the epiphyseal ends of long tubular bones and has a typical radiological presentation with well-defined borders and lytic subchondral lucency. 3 The reported frequency of these tumors ranges from 4 to 20% depending on the series reviewed.4–7 The histological variants of this neoplasm are conventional, and associated with an aneurismal bone cyst. Histological grading of GCT comprises grades I, II, and III. 8 Grade III tumors have giant multinucleated cells alternating with a sarcomatous stroma. Malignant GCT are classified as follows: i) primary; ii) secondary (when there is a history of a previously documented benign GCT), or iii) dedifferentiated. The difference between malignant GCT and the dedifferentiated malignant GCT is that the former has giant multinucleated cells alternating with stromal cells with sarcomatous morphology (Jaffe's histological grade III), whereas the latter is a benign GCT (Jaffe's grade I) juxtaposed with a sarcomatous tumor (osteosarcoma, malignant fibrous histiocytoma) and, as noted by Meiss et al., 9 there is no gradual transition between the two components, but rather an abrupt change, resulting in its characteristic bimorphic histologic appearance.
In this report, we present the case of a 30-year-old woman with a dedifferentiated GCT that affected the proximal radius.
Case Report
A 30-year-old woman was referred to our hospital with a history of pain and an increased volume in her right elbow which had been for 5 months. On physical examination, a 6-cm firm mass was detected in the lateral face of the elbow. The mass was painful, non-mobile, and attached to the deep planes corresponding to the proximal radial head. There was hypoesthesia and the flexion/extension and prono-supination movements of the patient's right arm were limited.
X-ray revealed a lytic lesion localized in the proximal meta-epiphysis of the radius with ill-defined borders and a permeative pattern of bone destruction. The lesion destroyed the lateral aspect of the radius's cortical. However, the humerus and the ulna the joint surface of the radius were uninvolved (Figure 1A).
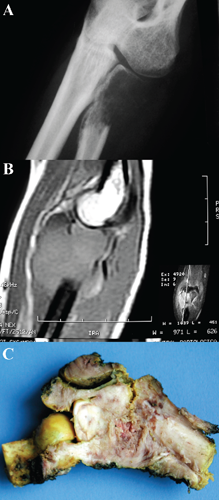
A) X-ray showing a lytic lesion of the proximal radial metaphysis that destroyed the lateral aspect of the radius's cortical. B) Sagittal T1 magnetic resonance imaging with fat saturation that showed radial lesion with uninvolved joint surface and peripheral spreading of the soft-tissue component. C) Radial tumor with a solid component in epiphysis and a fleshy metadiaphyseal component.
Sagittal magnetic resonance imaging showed a proximal radius lesion with periosteal reaction, destruction of the anterior cortical, and spreading to the adjacent soft tissues. The humerus, the ulna and the joint surface of the radius were uninvolved (Figure 1B).
After the clinical diagnosis of a primary bone tumor of the radius was confirmed, an en bloc resection was performed. The resected portion was a bone tumor of the distal radius with an extension to soft tissues. Macroscopically it consisted of two components: the first component was epiphyseal and didn't affect the joint cartilage of the radius and was located next to the bone with no extension to soft tissues. This component was solid and white in color with focal areas of yellowish discoloration. The second component of the tumor was more metadiaphyseal, had an extension to soft tissues, and a fleshy appearance with areas of hemorrhage and necrosis (Figure 1C). On histological examination, the epiphyseal tumor was subchondral, did not affect the articular cartilage and showed a GCT composed of mononuclear stromal cells alternating with giant multinucleated cells. The stromal cells were spindle-shaped with no nuclear atypia or hyperchromatism. In this stromal component, mitotic figures were not observed. The giant multinucleated cells were similar to osteoclasts, but with more nuclei arranged towards the center of the cell (Figure 2A). The remainder of the radial tumor was composed of a monotonous proliferation of small cells with rounded and basophilic nuclei and scant cytoplasm. The nuclei exhibited a finely-dispersed chromatin pattern. There were multiple areas of necrosis and the cells localized around vessels were generally preserved (Figure 2B). Scattered mitotic figures were identified, Homer-Wright- or Flexner-type rosettes were absent, and the periodic acid-Schiff preparation was negative in the cytoplasm of these cells. There was an interface, which consisted of spindle cells and necrotic tissue, between the GCT and the small, rounded, and basophilic component. Immunohistochemistry was negative for CD99, cytokeratin, and leukocyte common antigen, and was only positive for Vimentin. Due to the unusual histology, we consulted two bone tumor experts to discuss about this case. Their opinion was that this case was a dedifferentiated GCT with an undifferentiated, round cell, mesenchymal component. In the gammagraphic follow-up at 3 months after the en bloc resection, the patient presented multiple lesions in pelvis, sternum, right femur, and left supraclavicular.

A) Numerous osteoclastic giant cells located beneath the articular cartilage. B) An undifferentiated round cell tumor comprised the second component.
Discussion
The association of a low-grade tumor and a high-grade tumor is defined dedifferentiation in relation to bone tumors, 10 as in the case of dedifferentiated chondrosarcoma, 11 dedifferentiated parosteal osteosarcoma, 12 dedifferentiated adamantinoma, 13 dedifferentiated chordoma, 14 and dedifferentiated GCT. 9 The dedifferentiated giant-cell tumors are very rare. There are only four previously-reported and fully-documented cases in the literature. In these cases, the dedifferentiated component observed was a malignant fibrous histiocytoma, 9 a high-grade osteosarcoma, 15 and a low-grade fibroblastic osteogenic sarcoma. 16
The clinical implications of a conventional GCT and a dedifferentiated GCT are not the same. While the conventional GCT can be usually treated with curettage and bone grafting or en bloc resection, the dedifferentiated GCT requires chemotherapy, radiation therapy, and/or radical surgical resection.
Unfortunately, in the three cases reported and in also our case, the sarcomatous component was not included in the material of the initial biopsy and the patients were initially undertreated. This explains the importance of the site-of-biopsy and the amount of material harvested for the histological diagnosis. If the sarcomatous component is not observed, the patient will be undertreated, with clinical consequences. On the other hand, if the GCT component goes unnoticed, the patient will most likely be treated for a high-grade bone sarcoma without clinical consequences, but the peculiarity of the case (accompanied by secondary questions concerning the coexistence of a GCT with other bone tumors, which may lead to a better understanding of the nature of these lesions) would be missed.
We think that the association of a small round cell sarcoma and a GCT is not impossible and that it may even have a possible explanation, if we consider two previous observations. The first one provided by Scotlandi et al. 17 in which they demonstrated the presence of Ewing's sarcoma/FLI-1 hybrid transcripts in GCT. 17 The second observation reported that cells derived from the neuroblastoma (morphologically small round cells) express insulin-like growth factor 2 and promote osteoclastogenesis by this route. 18
Conclusions
To our knowledge, this is the first case reported in the literature of a dedifferentiated giant-cell tumor of the bone in which the dedifferentiated component is a small, round cell sarcoma.
Although very rare, the dedifferentiated GCT must be considered within the differential diagnosis of expansive and lytic bone lesions.