
Abstract
Hemorrhagic stroke is a devastating disease that lacks effective therapies. In the present investigation, we tested 6-bromoindirubin-3′-oxime (BIO) as a selective glycogen synthase kinase-3β (GSK-3β) inhibitor in a mouse model of intracerebral hemorrhage (ICH). ICH was induced by injection of collagenase IV into the striatum of 8- to 10-week-old C57BL/6 mice. BIO (8 μg/kg, IP) was administered following either an acute delivery (0–2 h delay) or a prolonged regimen (every 48 h starting at 3 days post-ICH). At 2 days post-ICH, the acute BIO treatment significantly reduced the hematoma volume. In the perihematoma regions, BIO administration blocked GSK-3β phosphorylation/activation, increased Bcl-2 and β-catenin levels, and significantly increased viability of neurons and other cell types. The prolonged BIO regimen maintained a higher level of β-catenin, upregulated VEGF and BDNF, and promoted neurogenesis and angiogenesis in peri-injury zones at 14 days after ICH. The BIO treatment also promoted proliferation of neural stem cells (NSCs) and migration of nascent DCX+ neuroblasts from the subventricular zone (SVZ) to the lesioned cortex. BIO improved functional outcomes on both the neurological severity score and rotarod tests. The findings of this study corroborate the neuroprotective and regenerative effects of BIO and suggest that the Wnt/GSK-3β/β-catenin pathway may be explored for the treatment of acute or chronic ICH.
Keywords
Introduction
Intracerebral hemorrhage (ICH), also known as hemorrhagic stroke, is a devastating brain disorder that accounts for approximately 10% of all stroke cases. ICH is a more emergent stroke subtype because it is more likely to result in fatality or severe disability in survivors1–4. Currently, no effective therapies exist to improve the prognosis following ICH 5 . Clinical management is principally conducted to minimize injury and to stabilize individuals in the perioperative phase. Surgical treatment to remove the hematoma could limit mass compression and injury from blood toxin accumulation, but the procedure remains controversial due to its invasive nature6,7. Also, although neuroprotectants and vascular protectants are beneficial for treating ICH in experimental animal models8,9, their translation into clinical applications have so far not been successfully validated.
Neural stem cells (NSCs) in the subventricular zone (SVZ) and subgranular zone (SGZ) give rise to neuroblasts that primarily migrate to the olfactory bulb and the hippocampal granular layers to integrate with local neural networks10,11. ICH stimulates cell proliferation and migration from the SVZ to the injured tissue12,13. Utilization of the endogenous regenerative mechanism after ICH is an attractive idea, but this potential treatment has rarely been explored.
The Wnt/β-catenin pathway plays a key role in regulating adult neurogenesis 14 . Wnt ligands bind to the frizzled (FZD) receptors, which, together with activation of its coreceptor low-density lipoprotein receptor-related protein (LRP), leads to destabilization of the complex containing axin, adenomatous polyposis coli (APC), and glycogen synthase kinase-3β (GSK-3β). This cascade results in the accumulation of β-catenin and its translocation into the nucleus to activate transcription factors for gene expression. As a critical inhibitor of β-catenin, GSK-3β serves as a viable target for modulation of the Wnt/β-catenin system. GSK-3β is ubiquitously expressed in the brain and is involved in many biological processes, including the survival, proliferation, migration, differentiation, polarity, and plasticity of neuroblasts and/or neurons 15 . 6-Bromoindirubin-3′-oxime (BIO) is a cell-permeable selective inhibitor of GSK-3β derived from Tyrian purple indirubins 16 . Inhibition of GSK-3β results in greater proliferation and asymmetric division of NSCs in the SVZ niche17–19. GSK-3β blockade has been shown to confer cytoprotective effects, which could ameliorate cell death following ICH20,21. This type of treatment is particularly effective in treating the multifaceted pathologies following ICH, in which cell death involves various triggers such as edema, ischemia, inflammation, etc.22,23. GSK-3β inactivation may also regulate neuronal differentiation of neural progenitors to promote neural regeneration. In this study, we evaluated whether BIO could provide dual roles of augmenting neuroprotection and neural regeneration, thereafter improving functional recovery after hemorrhagic stroke.
Materials and Methods
Animals and Intracerebral Hemorrhage Model
All experimental and surgical procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University and met National Institutes of Health (NIH) standards. C57BL/6 mice (8–10 weeks old, 25–28 g; Charles River Laboratories, Kinston, N Y, USA) were used in the study. Animals were housed in pathogen-free animal facilities and had free access to food and water.
The ICH procedure was performed as previously published 24 . Briefly, mice were anesthetized by intraperitoneal (IP) injection of pentobarbital (100 mg/kg). A scalp incision was made along the midline, and the stereotactic manipulator arms were adjusted such that the 25-gauge needle was positioned 0.2 mm [anterior–posterior (AP)] and 2 mm [right, medial–lateral (ML)] from the bregma. At these coordinates, a 1-mm cranial burr hole was drilled, and the needle tip was advanced 3.7 mm ventrally through the burr hole [from pia, dorsal–ventral (DV)]. Collagenase IV (0.075 U; Sigma-Aldrich, St. Louis, MO, USA) dissolved in 5 μl of phosphate-buffered saline (PBS) was injected gradually for 2.5 min (at a rate of 2 μl/min). The needle was left in position for an additional 10 min before withdrawing it at a rate of 1 mm/min. The incision was sealed by surgical glue (3M Corp., St. Paul, MN, USA). Body temperature was maintained at 37+0.5°C using a homeothermic blanket control unit (Harvard Apparatus Limited, Cambridge, UK).
Experimental Groups and Drug Delivery
Mice subjected to ICH were randomly divided into BIO and vehicle treatment groups. BIO (Stemgent, Lexington, MA, USA) was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) and then 0.9% saline for injection. For acute experiments investigating early cell death, stroke mice received BIO (8 μg/kg, IP) either at the time of ICH surgery (cotreatment) or with a 2-h delay after ICH and were sacrificed at 2 days after ICH. For chronic experiments investigating regenerative mechanisms, BIO was administered starting at 3 days post-ICH and continuously every 48 h until sacrifice. The day 3 time point was chosen based on the tissue loss information in the current and our previous study 25 . For sham and vehicle groups, mice received equivalent DMSO dissolved in saline. To detect proliferating cells, mice received daily IP injections of 50 mg/kg 5-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich) starting from day 3 after surgery.
Western Blot Analysis
Fresh brain tissue was isolated under a microscope from the peri-infarct region. Western blot analysis was performed to analyze protein expression following previous procedures 26 . In brief, brain tissue was lysed with modified radioimmunoprecipitation assay buffer [50 mmol/L HEPES (pH 7.3), 0.1% sodium deoxycholate, 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L sodium orthovanadate, 1 mmol/L NaF] and protease inhibitor cocktail (Sigma-Aldrich) with continuous homogenization. After 30 min, lysate was spun at 17,000 rpm for 15 min at 4°C, and supernatant was collected. Protein concentration was determined using a bicinchoninic acid (BCA) assay kit (Sigma-Aldrich). Equal amounts of protein (30 μg) were electrophoresed on 6% to 20% sodium dodecyl sulfate-polyacrylamide gradient gel (SDS-PAGE) in a Hoefer Mini-Gel system (Amersham Biosciences, Piscataway, NJ, USA) and transferred in the Hoefer Transfer Tank (Amersham Biosciences) to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA, USA). After blocking with 5% bovine serum albumin (BSA; Sigma-Aldrich) in Tris-buffered saline Tween 20 (TBST) buffer (20 mmol/L Tris, 137 mmol/L NaCl, and 0.1% Tween 20; Sigma-Aldrich) for 1 h, the PVDF membranes were incubated at 4°C overnight with primary antibodies against β-actin (1:5,000; Sigma-Aldrich), GSK-3β (1:1,000; Cell Signaling Technology, Danvers, MA, USA), phosphorylated GSK-3β (Y216) (1:1,000; Abcam, Cambridge, England, UK), β-catenin (1:500; R&D Systems, Minneapolis, MN, USA), Bcl-2 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), VEGF (1:500; Santa Cruz Biotechnology), or BDNF (1:500; Santa Cruz Biotechnology). Membranes were then washed with TBST three times and incubated with alkaline phosphatase-conjugated secondary antibodies (anti-rabbit or anti-mouse; 1:2,000; Promega, Madison, WI, USA) for 1 h at room temperature. After three washes with TBST and one wash with Tris-buffered saline (TBS; 20 mmol/L Tris, 137 mmol/L NaCl), blots were developed with bromochloroindolyl phosphate/nitroblue tetrazolium (BCIP/NBT) solution (Sigma-Aldrich).
Neuronal Cultures and Oxygen–Glucose Deprivation (OGD)
Primary cortical neuronal cell cultures were used in this test. The cerebral cortex was isolated from C57BL/6 fetal mice (E14–16) as previously described
27
. Briefly, embryos were harvested acutely from pregnant female mice sacrificed by isoflurane anesthesia and cervical dislocation. Embryos were decapitated, and the cortex was dissected under an anatomical microscope. Dissociated cortical neurons using trypsin were plated on 24-well plates (Corning, Corning, NY, USA) in Neurobasal media (Invitrogen, Carlsbad, CA, USA) supplemented with 2% B-27 (Invitrogen) and
Cell Death Assays
At selected days, cells cultured in 24- or 48-well plates were treated with OGD and different concentrations of BIO. LDH concentration and MTT activity were measured using Cytotoxicity Detection Kit reagents (Hoffmann-La Roche, Basel, Switzerland) and a microplate spectrophotometer (Bio-Tek Instruments, Winooski, VT, USA), respectively, according to the company's protocols.
Terminal Deoxynucleotidyl Transferase 2′-Deoxyuridine 5′-Triphosphate (dUTP) Nick-End Labeling (TUNEL) Staining
A TUNEL assay kit (Invitrogen) was used to examine cell death by detecting fragmented DNA in 10-μm-thick coronal fresh frozen sections as described previously 28 . Briefly, the fresh frozen sections were fixed with 10% buffered formalin (Sigma-Aldrich) for 10 min and then in ethanol/acetic acid (2:1) solution for 5 min and permeabilized in 0.2% Triton X-100 solution (Sigma-Aldrich), followed by incubation in equilibration buffer for 10 min. A recombinant terminal deoxynucleotidyl transferase (rTdT) and nucleotide mixture was then added on the slide at 37°C for 60 min in the dark. Reactions were terminated by 2× saline–sodium citrate (SSC) solution for 15 min. Immunohistochemistry was carried out for neuronal nuclei (NeuN; 1:300). Nuclei were counterstained with Hoechst 33342 (1:20,000; Molecular Probes, Eugene, OR, USA) for 5 min. TUNEL-positive areas within the hematoma were considered hematoma regions in our investigation, and the boundaries were used to determine the perihematoma regions under fluorescence microscopy.
Immunofluorescence Staining and Cell Count Quantification
Mice were sacrificed with an overdose of pentobarbital (100 mg/kg, IP; Sigma-Aldrich) at day 14 after stroke. Immunofluorescence staining was performed as described previously 26 . Briefly, frozen brain sections (10-μm thickness) or fixed sections [14-μm thickness; transcardiac perfusion with 4% paraformaldehyde (PFA) followed by postfixation] were processed and subsequently incubated with the primary antibodies including mouse anti-NeuN (1:200; Millipore, Billerica, MA, USA), rat anti-BrdU (1:200; Bio-Rad Laboratories), rabbit anti-glucose transporter 1 (GLUT1; 1:400; Millipore), and goat anti-doublecortin (DCX) (1:50; Santa Cruz Biotechnology) antibodies overnight at 4°C. The following day, sections were washed three times with PBS and then incubated with the secondary antibody cyanine 3 (Cy3)- or Cy5-conjugated anti-mouse, anti-rabbit, or anti-rat (1:400; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or Alexa Fluor 488 anti-goat (1:200; Abcam) for 1 h at room temperature. After three washes with PBS, all nuclei were stained with Hoechst 33342 (1:20,000; Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA) for 5 min as a counterstain. Sections were mounted with Vectashield fluorescent mounting medium (Vector Laboratories, Burlingame, CA, USA) and covered with glass coverslips for fluorescence microscopy (BX61; Olympus, Tokyo, Japan). Cell counting was performed following the principles of design-based stereology 29 . For each mouse, six 10- or 14-μm-thick sections spanning the entire region of interest were randomly selected for counting, and each of them was at least 100 μm apart from the next. Counting was performed on six randomly selected nonoverlapping fields per section located in perihematoma regions. Sections from different mice represent the same area in the anterior–posterior direction.
Neurological Severity (NSS) Test
A 10-point NSS was applied to estimate the neurological deficits at day 7 and day 14 after stroke according to published protocols 24 . The score consists of 10 parameters, including tasks on motor function, alertness, balance, and physiological behavior. One point was given for failure of the task and no point for success, with a maximal deficit score of 10.
Rotarod Test
The rotarod test of motor coordination and balance was performed at day 7 and day 14 after stroke 30 . Three days before, stroke mice were trained to walk on an accelerating rotating rod that increased in rotational speed from 4 to 40 rpm over the span of 5 min. The mean value of the last three training trials served as a preoperative baseline. Mice that were unable to remain on the rod for >90% of the total time during the last day of training were excluded from future tests.
Statistical Analysis
GraphPad Prism software (GraphPad, San Diego, CA, USA) was used for statistical analysis. Comparisons between two groups were performed using Student's t-test. One-way analysis of variance (ANOVA) analysis followed by Bonferroni correction was used for multiple-group comparisons. NSS score was analyzed using Mann–Whitney test for two group comparisons. For repeated measurements, two-way ANOVA was applied followed by Bonferroni correction. All data were shown as mean ± standard error of the mean (SEM). Statistical significance was defined as a value of p <0.05.
Results
Effects of BIO on GSK-3β and Hematoma Formation in the Brain
BIO is a specific inhibitor of GSK-3β that prevents phosphorylation at the Y216 residue, which would otherwise facilitate the activity of GSK-3β 16 . At higher concentrations, BIO is also a pan-janus kinase (JAK) inhibitor and blocks phosphorylation of signal transducer and activator of transcription 3 (STAT3) 31 . Limited information is available about in vivo dosage of BIO in animal experiments. A previous study using a dose of 50 mg/kg BIO found that the dosage was considered to be high and had some cytotoxic effects 32 . BIO was also given at low milligram per kilogram dosages for repeated IP administrations 33 . To identify a suitable dosage for BIO to produce sufficient GSK-3β inhibition in mice, total GSK-3β level and Y216 in the mouse brain were measured using Western blot analysis 48 h after IP injection of 0.08, 0.8, and 8 μg/kg BIO (Fig. 1). According to this assay, total GSK-3β levels remained relatively unchanged, while the phosphorylation of GSK-3β at the Y216 residue was largely inhibited by 8 μg/kg BIO (Fig. 1A–C). Furthermore, in ICH mice receiving different dosages of BIO, the hematoma volume was significantly reduced by the IP administration of 8 μg/kg BIO (Fig. 1D). We therefore implemented the dosage of 8 μg/kg in the following experiments.

Inhibitory effect of BIO on GSK-3β in the mouse brain. (A-C) Western blot analysis of 6-bromoindirubin-3′-oxime (BIO) target glycogen synthase kinase-3β (GSK-3β) was performed using tissue lysate from the perihemorrhagic regions following mouse intracerebral hemorrhage (ICH). (A) Representative immunoblots of all groups are shown. (B) Quantification of immunoblot intensities of total GSK-3β. (C) Quantification of immunoblot intensities of phospho-GSK-3β (Y216 residue). (D) Comparison of relative volumes of hematoma (mm3) in ICH animals treated with BIO. Mean+standard error of the mean (SEM). ∗p < 0.05 compared to ICH-only group. n = 6–8 per group.
BIO Affected GSK-3β Downstream Signaling β-Catenin and other Factors in the ICH Brain
Western blot analysis was conducted to measure several related proteins downstream of the GSK-3β pathway. GSK-3β activation inhibits cytoplasmic accumulation of β-catenin. At 24 h after stroke, Western blot demonstrated that ICH caused a downregulation of β-catenin signaling. BIO cotreatment (8 μg/kg, IP) or 2-h delayed treatment showed elevated β-catenin levels and increased the anti-apoptotic gene Bcl-2 (Fig. 2A and B). At 14 days after stroke, we again observed higher levels of β-catenin as well as the growth factors VEGF and BDNF, suggesting a long-lasting upregulation of the β-catenin pathway and regenerative factors (Fig. 2C and D).
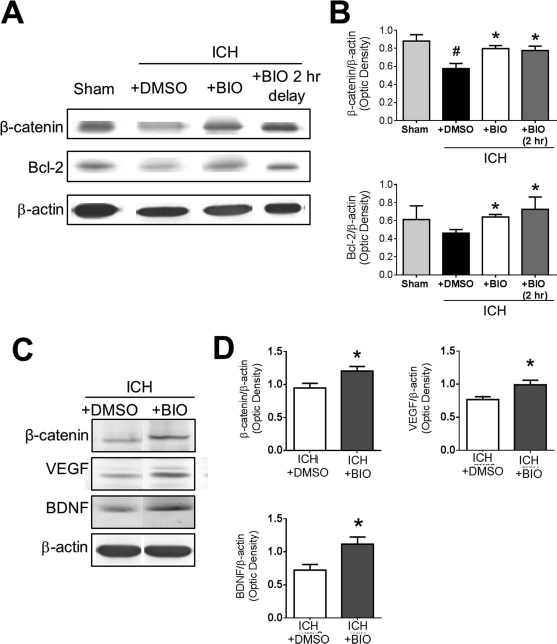
Regulatory effects of BIO on β-catenin expression and other factors in the ICH brain. (A) Representative Western blot images of perihematoma tissue lysates collected at 2 days post-ICH. (B) Quantification of immunoblots of β-catenin and the antiapoptotic factor B-cell lymphoma 2 (Bcl-2). Samples are normalized to β-actin loading control, and groups are normalized to sham. (C) Representative Western blot data of perihematoma tissue lysates at 14 days post-ICH to evaluate long-term BIO effects. (D) Quantification of immunoblot intensities of β-catenin, vascular endothelial growth factor (VEGF), and brain-derived neurotrophic factor (BDNF). Samples are normalized to β-actin loading control, and groups are normalized to ICH + dimethyl sulfoxide (DMSO) group. Mean+SEM. ∗p < 0.05 compared to ICH + DMSO group. #p < 0.05 compared to sham. n = 3 mice for sham group and n = 6–8 for other groups.
Effects of BIO on Cell Death In Vitro and In Vivo
Since BIO increased the antiapoptotic gene Bcl-2 and other presurvival/growth factors, we evaluated the BIO effect on cell death of cultured cortical neurons subjected to 3-h OGD. BIO (0.1, 1, or 10 μmol/L) was coapplied with OGD. Cell death was assessed using the LDH and MTT assays 24 h after OGD. BIO application significantly decreased LDH release and increased cell viability in a dose-dependent manner (Fig. 3A and B).
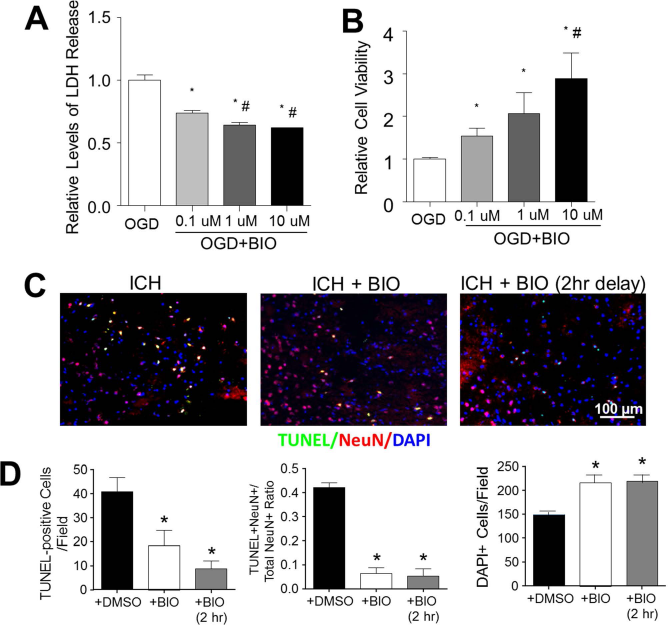
BIO-enhanced viability of neurons in vitro and in the ICH brain. (A) Lactate dehydrogenase (LDH) release correlated with neuronal cell death following oxygen-glucose deprivation (OGD). A dose-dependent neuroprotective effect was observed in BIO-treated cultures. (B) Relative cell viabilities determined by the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay in OGD with and without BIO treatments. Mean+SEM. ∗p < 0.05 compared to OGD + DMSO group. #p < 0.05 compared to OGD + 0.1 μM BIO group. (C) Representative images of multiplexed immunofluorescence of terminal deoxynucleotidyl transferase 2′-deoxyuridine 5′-triphosphate (dUTP) nick-end labeling (TUNEL, green), neuronal nuclei (NeuN, red), and 4′,6-diamidino-2-pheylindole (DAPI, blue). (D) Quantification of stereological counts of total cell numbers (DAPI+), numbers of cell death (TUNEL+ cells), and numbers of neuronal cell death (TUNEL+/NeuN+ colabeled cells, yellow signal). Mean + SEM. ∗p<0.05 compared to ICH + DMSO group. n = 4–5 culture batches/group.
In this animal study of ICH-induced brain cell death, ICH stroke mice received BIO cotreatment (8 μg/kg, IP) upon ICH or 2 h after the induction of ICH. Two days post-ICH, TUNEL immunohistochemical analysis on brain sections showed that both treatment paradigms significantly reduced the number of TUNEL-positive cells (neuronal and nonneuronal cells) and neuronal cell death (TUNEL+/NeuN+ cells). As a result, there were increased numbers of total surviving cells in the perihematoma region (Fig. 3C and D).
BIO Enhanced Neurogenesis and Angiogenesis in Perihematoma Regions
At 14 days after ICH, neurogenesis and angiogenesis in perihematoma regions were measured using immunofluorescence staining of newly generated (BrdU+) neurons and endothelial cells, respectively. The proliferation marker BrdU (50 mg/kg, IP) was injected daily from 3 days after stroke. In the perihematoma region, newly generated neurons were identified by double staining of NeuN and BrdU. The BIO treatment (8 μg/kg, I P, every 48 h from 3 days after stroke) significantly increased NeuN+/BrdU+ cells compared to the ICH control group (Fig. 4A–C). In ICH mice receiving BIO injections, there was a greater number of GLUT1+/BrdU+ cells (Fig. 5A–C). No NeuN+/BrdU+ or GLUT1+/BrdU+ double-positive cells were observed in similar cortical regions in the sham group (Figs. 4 and 5).

BIO-enhanced neurogenesis in the perihematoma region at 14 days after ICH. (A) Immunofluorescence staining showed 5-bromo-2′-deoxyuridine (BrdU, red) and NeuN (green) colabeled cells (white arrow) in sham, vehicle group, and BIO treatment group. (B) Three-dimensional image confirmed BrdU and NeuN colabeling. (C) Quantification data for NeuN+/BrdU+-colabeled cell counting. BIO significantly increased the numbers of NeuN+/BrdU+-colabeled cells in the perihematoma region compared to the control group. NeuN: green; BrdU: red; Hoechst 33342: blue. Mean + SEM. ∗p < 0.05. n = 8–12 animals/group.
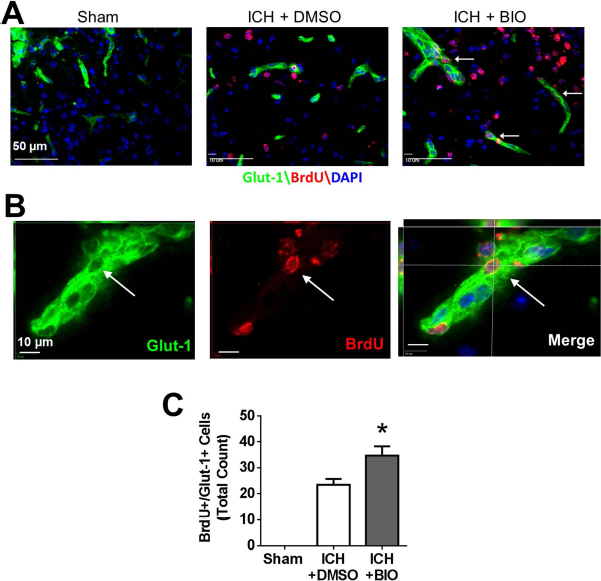
BIO-enhanced angiogenesis in perihematoma regions at 14 days after ICH. (A) Immunofluorescence staining showed BrdU (red) and glucose transporter 1 (GLUT1, green) colabeled cells (white arrow). (B) Three-dimensional image confirmed BrdU- and GLUT1-colabeled cells. (C) Quantification of GLUT1+/BrdU+-colabeled cells. BIO treatment resulted in greater numbers of GLUT1+/BrdU+-colabeled cells in the perihematoma region compared to the control group. GLUT1: green; BrdU: red; DAPI: blue. ∗p < 0.05. Mean ± SEM. n = 8–12 animals/group.
BIO Increased NSCs in the SVZ and Neuroblast Migration
We next examined proliferation of NSCs in the SVZ, as well as neuroblast migration. At 14 days after stroke, BrdU fluorescence showed proliferation of neuroblasts positively stained with DCX, and the BIO treatment significantly increased DCX+ and DCX+/BrdU+ cells compared to the vehicle treatment group (Fig. 6). These cells formed a migrating path from the SVZ toward the injured cortex, and the number of migrating cells (DCX+ cells) was enhanced by BIO (Fig. 6A–C).

BIO-enhanced proliferation and migration of neuroblasts from the SVZ. (A) Immunofluorescence staining for doublecortin (DCX, green), BrdU (red), and DAPI (blue). (B–D) Quantification for DCX+, BrdU+, and DCX+/BrdU+ colabeling. BIO treatment resulted in greater numbers of DCX+ cells, proliferating cells (BrdU+ cells), and newly divided migratory neuroblasts (DCX+/BrdU+). Mean ± SEM. ∗p < 0.05. ∗∗p < 0.01. n = 8–12 animals/group.
BIO Improved Functional Recovery After ICH
We used a modified NSS score and the rotarod test to evaluate functional recoveries after stroke. At 7 days after stroke, mice that received the BIO treatment had a much lower NSS score (Fig. 7A). Aside from the NSS score, the functional improvements associated with BIO were also corroborated using a separate behavioral test. In the rotarod test, BIO-treated mice were able to remain longer on the accelerating beam compared to vehicle mice at both 7 and 14 days after ICH (Fig. 7B).

BIO-improved functional recovery after ICH. (A) Neurological severity (NSS) evaluation showed moderate neurological recovery at 3, 7, and 14 days after surgery, with significant improvement at 7 days. (B) Rotarod test at 3, 7, and 14 days after ICH. BIO significantly improved the motor function at 7 and 14 days. Mean+SEM. ∗p < 0.01. ∗∗p < 0.05. n = 12–16 animals/group.
Discussion
The present investigation focuses on promoting endogenous regenerative activities mediated by pharmacological blocking of GSK-3β in the Wnt/β-catenin pathway. BIO is known to be a selective GSK-3 inhibitor with higher specific activity than lithium 34 . We show that BIO at low dosages (several hundred times below previously reported in vivo dosages) was able to markedly inhibit GSK-3β phosphorylation/activation in the mouse brain. Consistently, we observed increased β-catenin signaling and upregulations of several prosurvival/proregenerative factors in the post-ICH brain. The BIO treatment (either together with the ICH insult or with a 2-h delay) attenuated cell death, enhanced NSC proliferation and neuroblast migration, and increased the number of newly formed neurons and endothelial cells in the perihematoma region. The increased regenerative activities and improved functional recovery after stroke by BIO substantiates the potential therapeutic effects of this treatment.
Wnt signaling is critical for neurogenesis, neurite outgrowth, axon remodeling, synaptic plasticity, and neuronal morphogenesis in the adult brain35,36. Wnt inhibits GSK-3β and promotes β-catenin nuclear translocation to activate transcription of many genes related to cell survival, mitosis, and differentiation35,36. Downregulation of GSK-3β can increase the number of newborn neural cells 37 . We aimed to evaluate the therapeutic effects of targeting the Wnt/GSK-3β/β-catenin pathway. We postulated that BIO would enhance β-catenin signaling to promote cell survival and regeneration. The cytoprotective effects of BIO were shown in both the in vitro OGD model and the animal model of ICH. In the post-ICH brain, we identified that the β-catenin protein level declined in the perihematoma regions after ICH compared to sham controls, suggesting a negative impact on natural regenerative activities. There may be other signaling mechanisms, such as the AKT (protein kinase B) and mammalian target of rapamycin (mTOR) pathway, that are upregulated to compensate for the disrupted β-catenin signaling after ICH 38 . The interplay between these signaling pathways may explain the increased NPC proliferation and migration after ICH12,13.
The Wnt/GSK-3β/β-catenin pathway also directly regulates the maintenance of the blood–brain barrier (BBB), a key interface that underlies the pathology of ICH. The canonical Wnt proteins are critical for proper formation of tight junctions during organ development 39 , and disruption in the Wnt pathway resulting in a breakdown of the BBB 40 . GSK-3β inhibition has recently been shown to protect against microvascular hyperpermeability in hemorrhagic shock 41 . In the present investigation, the restoration of β-catenin levels through GSK-3β inhibition ameliorated the hematoma volume during the subacute phase of ICH. It was unclear, however, whether the reduction in hematoma volume directly contributed to the enhanced neuronal resilience following ICH. While further experiments are necessary to empirically disentangle these two BIO-associated phenomena, we hypothesize that the hematoma volume plays an important role in neuronal viability due to the pathophysiological events that arise. Extravasation of blood into the brain parenchyma results in elevation of intracranial pressure, which is correlated with greater neuronal pathology due to disrupted blood flow and associated hypoxia, as well as altered cerebral perfusion pressure that can lead to global ischemia 42 . Thus, a reduction in the hematoma volume should suggest both an amelioration of BBB disruption and an abated elevation of intracranial pressure, both of which would promote neuroprotection. Furthermore, direct injection of exogenous whole blood without disrupting the BBB exacerbates neuronal death, suggesting that the presence of the hematoma itself contributes, at least in part, to the injury 43 .
These findings suggest that Wnt/GSK-3β/β-catenin has an integral role in maintaining and stabilizing BBB integrity, vascular pathology, and neuroprotection. The decline in β-catenin levels after ICH and an inversion after therapeutic treatment highlight this signaling pathway as a particularly appropriate target. Our findings also verified that low dosages of BIO are most appropriate in the treatment of mouse ICH model. Higher dosages of BIO could also be a pan-JAK inhibitor against kinases such as tyrosine kinase (TYK)2, JAK1, JAK2, and JAK3, which could produce adverse off-target effects. In addition, BIO could interact with cyclin-dependent kinase 5 (CDK5)/p35 and CDK2/cyclin A, which may also result in undesirable effects. The BIO-promoted neurogenesis and angiogenesis in the perihematoma region are consistent with its stimulating effect on proliferation and differentiation35,36. Endogenous neural repair takes place following both ischemic stroke and hemorrhagic stroke13,44,45. A human study has identified the enhanced neurogenesis and migration of neuroblasts into perihematoma regions after ICH 12 . However, endogenous neurogenesis is normally not sufficient to compensate for neuronal damage. This warrants the identification and evaluation of novel drugs, such as BIO, as a small molecule capable of crossing the BBB. VEGF and BDNF are critical for angiogenesis and neurogenesis and show therapeutic benefits46,47. In an intraventricular hemorrhage model, BDNF has been identified as one of the key protective molecules in regenerative activities and functional recovery of the central nervous system (CNS) 48 . In the present investigation, we show the effect of BIO in the activation of the Wnt/β-catenin system, which then leads to upregulation of VEGF and BDNF, as well as enhancement of neurovascular repair and functional recovery in a mouse model.
In preclinical studies, ICH models show hematoma expansion and growth, providing sufficient construct validity for investigations on therapeutic intervention and outcome predictions 49 . The multiple regimens for BIO treatment was chosen in order to assess distinct phenomena after ICH, with the acute treatment used to assess neuroprotection and the delayed treatment used to assess regenerative events. Despite the limited dosage of the acute regimen, we confirmed that the single injection was sufficient to maintain the GSK-3β inhibition, as confirmed by Western blot. The time point for the long-term regimen was determined on the basis of our previous study, which identified hematoma progression measured for 2 days post-ICH 29 . To maximize the regenerative potential of BIO, we wanted to begin treatment after the peak of the tissue loss and injury size. This led to the selection of the day 3 initial time point of treatment for the long-term regimen. For the long-term regimen, we showed a similar level of GSK-3β inhibition as with the acute regimen. In conclusion, the early treatment targeted neuroprotection, while the delayed treatment promoted chronic effects, such as endogenous brain repair. There remains potential for further investigation into combining both the acute and long-term regimen. However, while the acute regimen provides insight into the neuroprotective nature of BIO, it is not as translationally applicable given that BIO must be coadministered or administered within a very narrow window following ICH. For any future studies, it may be more relevant to focus studies within the delayed regimen. Furthermore, the effect of BIO on neuroinflammation is highly intriguing and warrants investigation. Recent preclinical studies using stem cells or drug treatments showed improved outcomes after ICH through immunomodulation 50 and reduction of inflammation51–53. GSK-3β is involved in microglia/macrophage polarization following ICH 54 ; thus, the effect of BIO on inflammation modulation after ICH should be examined as an additional mechanism of the BIO treatment.
Conclusion
BIO treatment at a low dosage shows neuroprotective and regenerative effects and improves functional recovery after ICH. The effects were mediated by GSK-3β inhibition and activation of the Wnt/β-catenin pathway. BIO could be a potential agent to provide the dual role of a neuroprotective and regenerative treatment for ICH patients.
Footnotes
Acknowledgments
This work was supported by the NIH (grants NS062097 to L.W., NS085568 to L.W./S.P.Y., and NS091585 to L.W.), VA National Merit (grant RX000666 to S.P.Y.), AHA Predoctoral Fellowships (grant 16PRE31230001 to J.Y.Z.) and Postdoctoral Fellowship (grant 15POST25710112 to Z.Z.W.), National Natural Science Foundation of China (grants 81371355/81500989/81671191 to Y.Z. and 81571104 to J.L.), and Beijing Natural Science Foundation (grant 7142045 to Y.Z.). We are grateful to Dr. Jin Hwan Lee for his valuable advice on animal behavior tests. The authors declare no conflicts of interest.