
Abstract
We made a white matter injury (WMI) model with mild hindlimb dysfunction by right common carotid artery occlusion followed by 6% oxygen for 60 min at postnatal day 3 (P3), in which actively proliferating oligodendrocyte (OL) progenitors are mainly damaged. To know whether this model is appropriate for cell therapy using OL progenitors, the pathological response to mild hypoxia–ischemia (H-I) in neurons and OL lineage cells and myelination failure were investigated along with gene expression analysis. In WMI model rats, coordinated motor function, as assessed by the accelerating rotarod test, was impaired. The dysfunction was accompanied by myelination failure in layers I–IV of the sensorimotor cortex. Although several oligo2-positive OLs stained positive for active caspase 3 in the cortex and white matter at 24 h after H-I, few NeuN-positive neurons were apoptotic. Argyrophil-III staining for damaged neurons revealed no increase in the number of degenerating cells in the model. Moreover, the total number of NeuN-positive neurons in the cortex was comparable to that of controls 7 days later. Retrograde labeling of the corticospinal tract with Fluoro-Gold revealed no significant loss of layer V neurons. In addition, no decrease in the numbers of cortical projecting neurons and layers V–VI neurons in both motor and sensory areas was observed. Interestingly, the numbers of inhibitory GABAergic cells immunoreactive for parvalbumin, calretinin, or somatostatin were preserved in the P26 cortex. Gene expression analysis at P5 revealed 98 upregulated and 65 downregulated genes that may relate to cell survival, myelin loss, and differentiation of OLs. These data suggest that impaired motor coordination was not induced by neuron loss but, rather, myelination failure in layers I–IV. As OL lineage cells are mainly damaged, this WMI model might be useful for cell-based therapy by replacing OL progenitors.
Introduction
Cerebral white matter injury (WMI) is a complex amalgam of destructive and developmental distur bances1–4 and is a common feature of hypoxic–ischemic (H-I) brain injury in premature infants. Cerebral WMI causes several functional impairments such as perceptual, motor, and cognitive dysfunction4–8.
Along with the progression in the neonatal intensive care and neuroimaging, a profound shift in the features of WMI was recently reported9–11. While the incidence of large necrotic lesions of cystic periventricular leukomalacia (cystic PVL) has markedly declined, a milder form of WMI characterized by focal or diffuse nondestructive lesions (noncystic PVL) is increasing 2 . In addition, recent neuropathology studies revealed a 34% incidence of microscopic necrosis that is poorly defined by MRI3,12,13. Histopathological features of three classes of MRI-defined chronic WMI were recently shown in a sheep WMI model where each type displayed unique astroglial and microglial responses that corresponded to distinct forms of necrotic or nonnecrotic injury: diffuse WMI (DWMI), focal necrotic WMI, and microscopic necrotic WMI3,14.
One of the central features of chronic WMI is myelination failure that can arise from degeneration of late oligodendrocyte (OL) progenitors (pre-OLs) at the acute phase of WMI 15 . Alternatively, myelination failure might be a potentially reversible process that is linked to arrested pre-OL maturation in chronic WM lesions16,17. At the cellular level, pre-OLs are known as the principal target of WMI because of their selective vulnerability to H-I15,18. O4-positive pre-OLs are susceptible to cell death by free radicals and glutamate receptor-mediated toxicity after H-I19–22.
Cell-based therapy with grafted cells may be a promising treatment strategy for cerebral WMI from the aspect of not only replacing lost cells but also providing neurotrophic substances23–25. OL lineage cells derived from embryonic stem cells (ESCs) have recently been used for transplantation in WMI 26 . Therefore, OL lineage cells that are induced from pluripotent stem cells (iPSCs) 27 may be an appropriate donor cell type for WMI as well, especially in DWMI, which is a milder nondestructive lesion with predominant pre-OL degeneration followed by myelination failure. To identify potential cell-based therapies, it is very important to use more appropriate animal models that readily depict the pathology of the disease/injury. We previously reported on a WMI rat model 28 where the high-risk period of human WMI (G28–32) corresponds to P2–P4, a time when pre-OLs are rich and involved in OL development during H-I29,30.
Although our rat model depicts most of the pathology in DWMI, it remains unclear whether selective damage of pre-OLs is linked to functional deficits. To understand the pathology in the injured sensorimotor cortex responsible for hindlimb dysfunction in the DWMI model, the response to a milder nondestructive injury in neurons and OL lineage cells and myelination failure were investigated. In addition, gene expression analysis was performed to further investigate changes in the brain.
In this study, we clearly showed that motor coordination was impaired without obvious neuronal damage in a DWMI rat model in which oligo2-positive cells at P3 were partially lost at the acute phase, and upper layer myelin disruption was shown at the chronic phase. Notably, this model rat might be an appropriate model to give the possibility of cell-based therapy in DWMI using OL progenitors, although survival and differentiation of OL lineage cells in vivo should be investigated in future experiments.
Material and Methods
DWMI Rat Model
Animal care was carried out according to the guidelines of the Institute for Experimental Animal Science at Nagoya City University Medical School (Nagoya, Japan). All experimental procedures were approved by the committee of animal experimentation of Nagoya City University Medical School, and every effort was made to minimize the pain and discomfort of the animals used in this study. Male neonatal Wistar rats (P3) were obtained from Japan SLC, Inc. Pups were housed with their mothers, which had access to standard chow and water ad libitum, under a constant 12-h dark/light cycle. A total of 92 pups were used for this experiment.
DWMI model rats were established according to a previously described method 28 . Briefly, P3 pups (n = 65) were subjected to right common carotid artery occlusion (RCCAO) under isoflurane (Pfizer, New York, NY, USA) anesthesia [5% (v/v) induction, 1.5% (v/v) maintenance]. After a 2-h recovery period with their dam, pups were exposed to 6% (v/v) O2 hypoxia for 60 min in a container submerged in a 38°C water bath. The pups were returned to their dam and maintained under normal conditions with a light/dark cycle of 12 h. Sham-operated rats (n = 27) were used as the control. The rats were allowed to wean at P25.
Behavioral Testing
Motor coordination was tested using the accelerating rotarod system31,32. For habituation, 8-month-old rats (DWMI: n = 16, sham operated: n = 12) were placed on a rotating rod (10 rpm) for 5 min for 3 successive days. For the test, rats were placed on an accelerating rotarod from 4 to 40 rpm in 60 s, three times. The maximal time that the rat was able to stay on the top of the rotating rod was used for data analysis.
Immunostaining
Rats were deeply anesthetized with pentobarbital (>50 mg/kg; Kyorituseiyaku, Tokyo, Japan) and perfused with 4% (w/v) paraformaldehyde (PFA; Sigma-Aldrich, St. Louis, MO, USA) in 0.1 M of phosphate-buffered saline (PBS). Brains were postfixed overnight followed by cryoprotection in 20% (w/v) sucrose in 0.1% (v/v) PB. Serial coronal sections (20 μm) were cut and processed for histology. For immunohistochemistry, endogenous peroxidase was inactivated with 0.6% (v/v) H2O2 (Wako, Tokyo, Japan) dissolved in PBS + 0.1% (v/v) Triton (Nacalai Tesque, Inc., Kyoto, Japan) for 30 min. After washing with PBS, sections were blocked with 3% (v/v) serum and incubated overnight at 4°C with primary antibodies. Sections were then reacted with biotinylated secondary antibodies (Vector Laboratories Inc., Burlingame, CA, USA) and visualized using the Vector ABC kit (Vector Laboratories) and 3,3′-diaminobenzidine (DAB; Dojindo, Kumamoto, Japan). For immunofluorescence, sections were incubated with 3% (v/v) normal goat serum and incubated overnight at 4°C with primary antibodies. Appropriate fluorophore-conjugated secondary antibodies (Molecular Probes, Inc., Eugene, OR, USA) were used for visualization.
The primary antibodies used were as follows: rabbit anti-olig2 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-NeuN (1:200; Millipore, Bedford, MA, USA), mouse anti-myelin basic protein (MBP) (1:1,000; Covance, Princeton, NJ, USA), adenoma polyposis coli (APC) (CC-1; 1:250; Millipore), rabbit anti-activated caspase 3 (1:200; Millipore), mouse anti-SATBII (1:200; Abcam, Cambridge, UK), rabbit anti-Ctip2 (1:200; Abcam), rabbit anti-Fluoro-Gold (1:3,000; Millipore), mouse anti-parvalbumin (1:1,000; Swant, Bellinzona, Switzerland), mouse anti-calretinin (1:1,000; Swant), and rat anti-somatostatin (1:250; Millipore). The secondary antibodies were as follows: biotinylated anti-rabbit IgG (1:220; Vector Laboratories, Inc.), biotinylated anti-mouse IgG (1:220; Vector Laboratories, Inc), goat anti-mouse Alexa 488 (1:500; Molecular Probes, Inc.), and goat anti-rabbit Alexa 594 (1:500; Molecular Probes, Inc.).
For cell counting, coronal sections (40 μm thick) at the level of the hindlimb-regulating cortex area (bregma: –2.04) were collected. Cells were counted in the motor and sensory areas in each section (400 μm × 400 μm). Following NeuN, Ctip2, SATBII, and GABAergic subtype staining, cells were counted in the motor and sensory areas (400 μm width, I to VI layers). For quantification of MBP immunostaining, MBP signal in the motor and sensory area (I–IV, V–VI layers: 350 μm × 350 μm each) was digitalized at 16 bit using ImageJ software from the National Institutes of Health (NIH, Bethesda, MD, USA). The appropriate intensity value was set as the threshold, and the pixel number above the threshold was considered positive staining. Discrimination of upper (I–IV) and deep (V–VI) layers was determined using Ctip2 (V–VI layer marker) immunostaining. The ratio of ipsilateral to contralateral pixel number is shown in the graphs.
Fluoro-Gold Labeling
At P20, the spinal cord was exposed at the C3–C4 level by laminectomy under anesthesia (n = 5). One microliter of 2% (w/v) Fluoro-Gold (FG; Biotium, Hayward, CA, USA) was injected into the center of the dorsal corticospinal tract (1-mm deep from the dura) with a glass micropipette (tip diameter: 60–80 μm). Rats were anesthetized and perfused with 4% (w/v) PFA 6 days later. Brain sections were cut for histological analysis. FG-labeled cells were detected using an anti-FG antibody. The cell numbers were counted in every eighth brain section in the hindlimb motor-regulating cortex [three sections for each brain, 700 × 700 μm2 in motor (M1 and M2) and sensory areas].
Argyrophil-III Silver Staining
Under deep anesthesia with pentobarbital, animals (DWMI: n = 5, sham operated: n = 4) were perfused with 0.1% (w/v) cacodylate (Wako) buffer solution containing 2% (w/v) PFA and 2.5% (w/v) glutaraldehyde (Wako). Brains were removed 24 h after perfusion and postfixed for 1 week at room temperature. Serial coronal sections (40 μm) were cut and processed for argyrophil-III staining. The sections were esterified in 1-propanol (Wako) containing 1.2% (v/v) sulfuric acid (Katayama Chemical., Osaka, Japan) and 2% (v/v) distilled water for 16 h. After rehydration in 50% and 25% (v/v) 1-propanol and distilled water, sections were processed in silicotungstate (Wako) physical developer as described previously33,34. For cell counting, the number of positive cells in the selected area (400 μm × 400 μm) was counted in the hindlimb-regulating cortex (DWMI 24 h: n = 4, DWMI 48 h: n = 4).
Statistics
Data were analyzed using StatView 5 software (SAS institute, Cary, NC, USA). All data are presented as mean ± SEM, and values of p < 0.05 were considered significant. Student's t-test was used for comparison between sham-operated and DWMI groups in rotarod test. Student's t-test was also used for comparison between contralateral and ipsilateral sides in the number of APC-positive cells, NeuN-positive cells, STABII-positive cells, Ctip2-positive cells, FG-positive cells, and GABAergic markers such as parvalbumin, somatostatin, and calretinin. For the comparison of the percentage of caspase 3-positive cells and NeuN-positive cells, one-way ANOVA followed by Scheffé post hoc test was used in four groups (sham-operated ipsilateral side, sham-operated contrarateral side, DWMI ipsilateral side, DWMI contralateral side). To detect statistical significance in the intensity of MBP staining, one-sample t-test was used for comparison as the intensity of contralateral side was estimated as onefold.
Results
Impaired Motor Coordination and Altered Myelination Were Induced in the DWMI Rat Model
To investigate sensorimotor function in our DWMI rat model, we performed the accelerating rotarod test at adulthood (8 months old). DWMI rats (n = 16) showed a significantly lower score compared with sham-operated animals (n = 12), indicating that motor coordination was impaired (Fig. 1A). In our analysis of motor deficit scores (beam walk ability test, hindlimb retract test, forepaw grasp test) 35 , DWMI rats showed mild functional deficits mainly in the left hindlimb (data not shown). Therefore, we focused on the hindlimb-regulating somatosensory cortex.

Coordinated motor dysfunction with myelination failure in the WMI model rat. (A) Motor coordination was assessed using the accelerating rotarod system. The DWMI group showed significantly lower scores compared with the sham group (DWMI: n = 16, sham: n = 12, *p < 0.05 in Student t-test). (B) Immunostaining of APC-positive cells (red) in the ipsilateral white matter with DAPI (blue). Scale bar: 50 μm. Number of APC-positive mature OLs decreased in ipsilateral WM at 6-month-old model rats (n = 4 *p < 0.05 in Student t-test). (C) Weaker MBP immunoreactivity was observed in the M1 and S1 areas of the ipsilateral cortex (2 months old). Scale bar: 100 μm. M1, primary motor cortex; M2, secondary motor cortex; S1, primary sensory cortex. (D, E) Quantification of MBP signal intensity at P26 (n = 5) (D) and P74 (n = 4) (E). Discrimination between upper (I–IV) and deep (V–VI) layers was determined by Ctip2 (V–VI layer marker) immunostaining (*p < 0.05 in one-sample t-test).
Disturbances to OL development after H-I were confirmed in the model by immunostaining to mature OL marker APC. Many cells that expressed APC in the cytoplasm were detected in the WM (Fig. 1B). There was a 16.0% reduction of APC-positive cells in the ipsilateral side of 6-month-old DWMI rats (n = 4) (Fig. 1B). To confirm whether this reduction in mature OLs coincided with a disturbance to myelination, the hindlimb-regulating sensorimotor cortex was immunostained using the MBP antibody. In the ipsilateral cortex, an obvious decline in MBP-positive myelin was observed compared with the contralateral cortex, which extended from the motor area to the sensory area (Fig. 1C). We next quantitated the intensity of MBP signal using ImageJ software in the upper (I–IV) and deep layers (V–VI) of the motor and sensory cortex at P26 (period of active myelination, n = 5) and P74 (period of myelination completion, n = 4). At both developmental stages, MBP intensity significantly decreased in the upper layer of the sensory area (Fig. 1D). There was a slight reduction in MBP signal detected in the upper motor area (P26, p = 0.054; P74, p = 0.07), while no change in MBP intensity was observed in the deep layers of the motor and sensory areas. These data indicate that P3 H-I leads to functional deficits with loss of mature OLs and altered myelination in the upper sensorimotor cortex.
H-I Causes Apoptotic Cell Death of Glial Cells, but not Neurons
To monitor cell death following H-I, we performed active caspase 3 (casp3) immunostaining at 24 and 48 h after injury. Casp3-positive cells were detected in sham-operated control brains (n = 3 for each time points) (Fig. 2A and B) as well as in the motor and sensory cortex of model rats at both time points (24 h: n = 6, 48 h: n = 4). However, no significant difference was observed between H-I and control rats (Fig. 2A and B). Interestingly, the number of casp3-positive cells increased in ipsilateral WM at 24 h compared with the contralateral side (Fig. 2A), indicating that the WM area was sensitive to H-I injury. Because other DWMI animal models have been shown to have more neuronal death after H-I compared to controls, neuronal death was investigated in our model. We monitored the numbers of casp3/NeuN double-labeled cells at 24 and 48 h. No double-labeled neurons were detected at either time point (Fig. 2C). Almost all of the casp3-positive cells were colocalized with olig2 (Fig. 2D) or glial fibrillary acidic protein (GFAP) (Fig. 2E), suggesting that glial cell death mainly occurred in cortical WM. No significant differences in the total number of NeuN-positive neurons were observed in the ipsilateral motor and sensory cortices at 48 h compared with the contralateral cortex (sham: n = 3, DWMI: n = 4) (Fig. 2F).
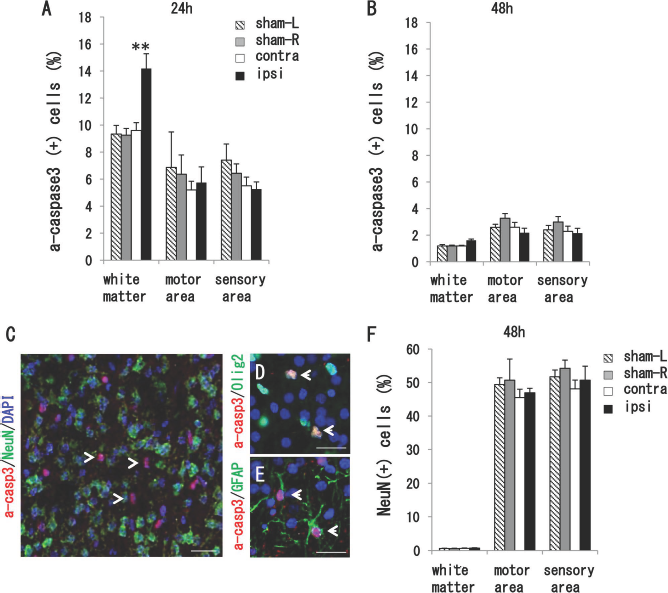
Apoptotic cell death was not evident in neurons after H-I. (A) Immunostaining of active caspase 3 (a-casp3) at 24 h and (B) 48 h after H-I. Although casp3-positive cells were detected in sham-operated control brains (n = 3), no significant difference was detected in the motor and sensory areas at each time point (24 h: n = 6, 48 h: n = 4). Notably, the numbers of positive cells increased in ipsilateral WM at 24 h compared with the contralateral side (A). *p < 0.05 in one-way ANOVA followed by Scheffé F-test. (C, E) Few a-casp3/NeuN double-positive apoptotic neurons were detected at 24 h. Almost all a-casp3-positive cells colocalized with olig2 (D) or GFAP (E). Arrows indicate double-positive cells. Nuclei were stained with DAPI (blue). Scale bars: 20 μm. (F) The percentage of NeuN-positive cells in WM and the cortex did not change between DWMI rats (n = 4) and the sham control (n = 3) at 48 h.
As argyrophil-III silver staining is known to detect degenerating neurons at very early phases 34 , we monitored the extent of degeneration using argyrophil-III silver staining at 24 h, 2, 3, and 6 days after injury (DWMI; n = 5, sham: n = 4). In the motor and sensory cortices, H-I did not induce neuronal degeneration (Fig. 3A and B). At 24 h and 48 h after injury, some dark cells with large cell bodies and processes were detected in the deep regions of the ipsilateral cortex (24 h: 13.5 ± 3.2, 48 h: 12.5 ± 3.3, n = 4) [Fig. 3D (inset) and E]. However, these cells were also observed in the contralateral side (24 h: 14.0 ± 4.5 cells, 48 h: 14.0 ± 3.7 cells, n = 4) (Fig. 3E) and sham-operated cortex at the same levels. There was no change in the expression pattern of silver-stained cells (damaged neurons) at days 3 and 6 (data not shown). Thus, apparent neuronal cell loss was not induced after H-I in the sensorimotor cortex in our model.

Detection of damaged cells at the very early phase using argyrophil-III staining. (A, B) Argyrophil-III silver staining was carried out at 24 h after H–I (DWMI: n = 5, sham: n = 4). Typical degenerating dark neurons were not detected in either the motor and sensory areas. (C, D) Some positively stained neurons with large cell bodies and processes were detected in the ipsilateral deep cortex (D, inset). However, a similar population of cells was also observed on the contralateral side (C) at 24 and 48 h. Scale bar: 100 μm in (A–D). Inset: Some positive neurons were detected on the contralateral and ipsilateral cortex. Scale bar: 25 μm. (E) Cell counting of argyrophil-III staining positive cells revealed that no significant difference in the number of the positive neurons between the contralateral or ipsilateral cortices at 24 h and 48 h after H–I (24 h: n = 4, 48 h: n = 4).
P3 H-I Injury Does not Affect Cortical and Corticospinal Projections Associated with Hindlimb Motor Function
To investigate whether H-I injury at D3 causes alteration of neural projections in the sensorimotor cortex, we stained cortical projecting cells with anti-SATB II, a marker of CC projecting neurons, and Ctip2, a marker of layers V–VI neurons (Fig. 4A). In the cerebral cortex, SATB II was mainly expressed in layer sII–IV, while some positive staining was detected in deep layers. Ctip2 was detected in layers V–VI (Fig. 4A, magnification). There was no significant difference in the number of SATB II-positive cells in the motor area between the contralateral and ipsilateral side (919 ± 72 cells vs. 955 ± 40 cells, n = 5) (Fig. 4C) as well as in the sensory area (1,082 ± 96 vs. 1,074 ± 66 cells, n = 5) (Fig. 4D). There was also no difference observed between Ctip2-positive cells in the motor area (408 ± 25 cells vs. 371 ± 14 cells, n = 5) (Fig. 4C) and in the sensory area (420 ± 31 vs. 377 ± 33 cells, n = 5) (Fig. 4D). The total number of NeuN-positive neurons in the contralateral and ipsilateral side was almost the same in the motor area (1,187 ± 82 cells vs. 1,170 ± 29 cells, n = 5) (Fig. 4C) and sensory area (1,466 ± 100 vs. 1,422 ± 63 cells, n = 5) (Fig. 4D).

Absence of cortical projection neuron loss in the WMI rat model. (A) SATBII-positive cortical projecting neurons (green) and Ctip2-positive layers V–VI neurons (red) were observed in the contralateral and ipsilateral sensorimotor area at P10. Staining revealed that most cells in layers II–IV were SATBII-positive, while Ctip2-positive projecting neurons were detected in layers V–VI. Scale bar: 50 μm. (B–D) Number of SATBII-positive, Ctip2-positive, and NeuN-positive cells in the motor cortex (MC) and sensory cortex (SC) (B). No significant differences in number were observed in the motor (C) and sensory (D) areas between the contralateral and ipsilateral side (n = 4–5).
To estimate the damage to corticospinal neurons in the cortex, the retrograde neuronal tracer FG was injected into the spinal cord at C3–C4 level at P20 and examined 6 days later. FG successfully incorporated into pyramidal neurons and was detected in layer V of the area from the forebrain to the hindbrain (Fig. 5A).

H-I does not induce abnormal corticospinal projection. (A, B) Immunostaining of Fluoro-Gold (FG) in the P26 cortex revealed positive cells at layer V in the hindlimb-regulating cortex. Scale bar: 100 μm. The number of FG-positive neurons did not change in either the motor or sensory area at P26 (n = 5) (B). (C, D) Immunostaining of parvalbumin, somatostatin, and calretinin. No significant differences in the number of each subtype were detected in the motor (C) or sensory (D) areas (n = 3–5).
The same number of FG-positive cells was detected in the hindlimb-regulating motor area of the ipsilateral and contralateral sides (245.2 ± 27.9 cells vs. 313.6 ± 26.4 cells, n = 5) (Fig. 5B) as well as in the sensory area (238 ± 27.5 cells vs. 290.6 ± 20 cells, n = 5) (Fig. 5B), indicating that the hindlimb-regulating corticospinal projection was intact after H-I. Furthermore, the number of corticospinal projecting neurons was maintained at 9 months after H-I (data not shown), suggesting that secondary loss of neurons was not induced by disturbances to myelination at the site of injury. As inhibitory neurons have been reported to decrease chronically and delay the development of these inhibitory neurons in a DWMI model36,37, we monitored the expression of inhibitory interneuron subtypes using antibodies against parvalbumin, somatostatin, and calretinin, (n = 3–5) (Fig. 5C and D). In the P26 rat cortex, no significant change in the number of each cell subtype was detected in the motor (Fig. 5C) and sensory areas (Fig. 5D).
H-I Alters Gene Expression in the White Matter and Sensorimotor Area
To assess the factors that contribute to myelination disturbances and motor dysfunction in our DWMI model, we performed cDNA microarray analysis in the P5 sensorimotor cortex (n = 2 for DWMI and sham operated) (Tables 1 and 2).
Upregulated Genes in DWMI Rat Model

Mmp28, matrix metallopeptidase 28; Adamts1, a disintegrin-like and metallopeptidase with thrombospondin type 1 motif, 1; Plau, plasminogen activator; C1r, C1r protein; Klk4, kallikrein-related peptidase 4; Klkb1, kallikrein B, plasma 1; Acr, acrosin prepropeptide; Capn8, calpain 8; Ace, angiotensin I-converting enzyme 1; Hk3, hexokinase 3; Igf2, insulin-like growth factor 2; Galm, galactose mutarotase; Kl, klotho; Pofut1, protein O-fucosyltransferase 1; Slc3a1, solute carrier family 3, member 1; Aqp1, aquaporin 1; Ednra, endothelin receptor type A; Cnr2, cannabinoid receptor 2; Fxyd1, FXYD domain-containing ion transport regulator 1; Slc37a4, solute carrier family 37 (glucose-6-phosphate transporter), member 4; Ccl2, chemokine ligand 2; Trpv4, transient receptor potential cation channel, subfamily V, member 4; Sod3, superoxide dismutase 3, extracellular; Tf, Serotransferrin precursor; Fads1, fatty acid desaturase 1; Fos, FBJ osteosarcoma oncogene; Fabp4, fatty acid-binding protein 4 adipocyte; Slc39a9, solute carrier family 39 (zinc transporter), member 9; Kcnj13, potassium inwardly rectifying channel, subfamily J, member 13; Kcne2, potassium voltage-gated channel, Isk-related subfamily, gene 2; Chrna6, cholinergic receptor, nicotinic, a polypeptide 6; Slc1a3, solute carrier family 1, member 3; Htr2c, 5-hydroxytryptamine receptor 2C; Sult1a1, sulfotransferase family 1A, phenol-preferring, member 1; Serpinc1; serine (or cysteine) peptidase inhibitor, clade C (antithrombin), member 1; Serpinb1a, serine (or cysteine) peptidase inhibitor, clade B, member 1a; Renbp, rennin-binding protein; Msx1, homeobox, msh-like 1; Hspb1, heat shock protein 1; Ppm1b, protein phosphatase 1B, magnesium dependent, β isoform; Styxl1, serine/threonine/tyrosine interacting-like 1; Eya2, eyes absent 2 homolog; Sulf1, sulfatase 1; Serpinc1, serine (or cysteine) peptidase inhibitor, clade C (antithrombin), member 1; Fabp4, fatty acid-binding protein 4, adipocyte; Itgb6, integrin β 6; Klkb1, kallikrein B, plasma 1; Slc3a1, solute carrier family 3, member 1; Ramp3, receptor (calcitonin) activity-modifying protein 3; Ttr, transthyretin gene; Degs2, degenerative spermatocyte homolog 2 (Drosophila), lipid desaturase; Sod3, superoxide dismutase 3, extracellular.
Downregulated Genes in DWMI Rat Model

Kcnj16, potassium inwardly rectifying channel, subfamily J, member 16; Cacna2d2, calcium channel, voltage-dependent, α 2/δ subunit 2; Hcn2, hyperpolarization-activated, cyclic nucleotide-gated K+ 2; Gabrg3, γ-aminobutyric acid (GABA) A receptor, subunit γ 3; Chrnb4, cholinergic receptor, nicotinic, β polypeptide 4; Clic5, chloride intracellular channel 5; Accn4, amiloride-sensitive cation channel 4, pituitary; Il2rg, interleukin 2 receptor, γ chain; Il7, interleukin 7; RT1-S3, RT1-149 protein; Oas1b, 2′-5′ oligoadenylate synthetase 1B; Olr121, olfactory receptor Olr121; Taar1, trace amine-associated receptor 1; Olr1507, olfactory receptor Olr1507; Olr482, olfactory receptor Olr482; Olr1375, olfactory receptor Olr1375; Olr491, olfactory receptor Olr491; Olr192, olfactory receptor Olr192; Olr434, olfactory receptor Olr434; Olr1350, olfactory receptor Olr1350.
Of the 163 genes that changed expression, 98 were upregulated, and 65 were downregulated in the ipsilateral sensory motor cortex compared with the contralateral cortex. Gene ontology (GO) analysis using GENE CODIS2.0 (http://genecodis2.dacya.uccm.es/) revealed that genes involved in the carbohydrate metabolic process, proteolysis, ion transport, negative apoptosis regulators, hypoxia-responsive factors, inflammatory response, and oxidation–reduction process were upregulated in the ipsilateral cortex (Table 1). Among the downregulated genes, many were related to ion transport and the G-protein-coupled receptor protein signaling pathway (Table 2). Many genes responsible for neuronal survival, myelin loss, and differentiation were also altered in the DWMI model.
Discussion
For basic science to cell-based therapies, it is very important to use more appropriate animal models that accurately depict the pathology of the disease. We previously reported on a DWMI rat model where the high-risk period of human WMI corresponds to P3, a time when O4-positive pre-OLs are rich and actively produced 28 . However, it remains unclear whether the pathology of the injury of OL lineage cells is related to the functional deficits in the DWMI model. To clarify the pathology in the injured sensorimotor cortex responsible for hindlimb dysfunction in the DWMI model, the response of neurons and OL lineage cells to a mild, nondestructive H-I injury was investigated. In addition, the effect on myelination and gene expression was analyzed. We clearly showed that motor coordination was impaired without obvious damage to neurons in the DWMI model in which olig2-positive cells were partially lost at the acute phase, and myelin disruption in layers I–IV was observed at the chronic phase.
Specific Feature of the DWMI Rat Model
The significant feature of our DWMI rat model was minor neuron damage compared with previous reports29,38,39. Distinct cell death of neurons was not shown in the sensorimotor cortex after H-I; however, CC1-positive mature OLs were decreased at adulthood. As described by previous reports, Fluoro-Jade (FJ) B-positive degenerating neurons were detected in deep layers (layer VI) 38 , an area vulnerable to neonatal H-I39,40. We also detected FJ B-positive cells at 24 and 48 h in deep layers (layers IV–V) in the DWMI model (data not shown). In contrast, no neuron damage was detected when we used the argyophil-III staining method to detect the very early phase of neuron damage 41 . Assessment of cell death using FG injection, STABII, Ctip2, and GABAergic marker labeling strongly confirmed no neuronal loss in the somatosensory cortex in the DWMI model. In addition, few casp3/NeuN double-positive apoptotic neurons were detected in the cortex at 24–48 h after the lesion. These data indicate that neuron damage in the somatosensory cortex of DWMI rats was minor following H-I and not aggressive enough to induce significant cell death.
The principal target in the acute phase of WMI is O4-positive pre-OLs because they are selectively vulnerable to H-I15,18–22,42. Three classes of MRI-defined WMI at the chronic phase have also recently been revealed in a sheep model of WMI: DWMI, focal necrotic WMI, and microscopic necrotic WMI3,14. In particular, DWMI is a major form of chronic WMI and is characterized by astrogliosis and myelination disturbances. Using histological assessment, casp3-positive apoptotic cells only increased in ipsilateral WM at 24 h after H-I (Fig. 2A and B), which were mostly olig2 and casp3 double-positive OL lineage cells and some reactive GFAP-positive cells. Reduced MBP staining in layers I–IV of the sensory cortex was revealed at 6 months (chronic phase) compared with the contralateral cortex. Furthermore, a small, but significant, reduction in CC1-positive mature OLs was detected in WM, while a transient increase in OPCs was observed (data not shown). These data suggest that both the cell death of OL lineage cells at the acute phase and OPC maturation arrest at the chronic phase may contribute to myelination failure in the upper cortical layers in adulthood. We also suggest that this WMI rat model may be an appropriate DWMI model as it depicts the major form of chronic WMI, including myelination disturbances in OL lineage cells and reactive responses in astrocytes.
Transient reduction of excitatory neurons in the cerebral cortex and prolonged decreases, or developmental delays, to inhibitory interneurons was reported in chronic hypoxia from P3 to P1136,37,43. In our DWMI model, no expression of casp3 was detected in NeuN-positive neurons at 24–48 h after H-I (Fig. 2A and B), and no reduction in the total number of neurons was observed in the P10 cortex (Fig. 2D). Notably, no decrease in the number of both excitatory neurons (retrogradely labeled FG-positive layer V cells) and inhibitory neurons (parvalbumin-, somatostatin-, calretinin-positive cells) was observed in the DWMI rat model following H-I at P3. Thus, neurons in the somatosensory cortex were not damaged by H-I in the DWMI rat model rather than the recovery after transient reduction or delays such as in chronic hypoxia. With respect to differences between our DWMI model and previously reported animal models, it is necessary to consider the differences in pathology caused by the experimental conditions such as the age of the animal and the duration of H-I.
Probable Mechanism for Functional Deficits
Although no neuronal loss in the sensorimotor cortex was observed, dysfunction to sensory/motor coordination was revealed using the accelerating rotarod test. One possibility for this functional deficit could be a disturbance to neuron–glia interaction as the intensity of myelin staining was lower in the upper layer in the DWMI brain (myelination failure). For example, myelin loss may change conduction velocity in the sensorimotor inputs–outputs and/or intracortical association, which leads to an imbalance in information processing for coordinated movement. It is possible that synaptic input from the hypothalamus and/or intracortical interaction between layers I–IV dendrites was affected in the DWMI model, resulting in dysfunction to sensory motor coordination.
Another possible reason for the functional deficits may be neuronal reorganization in the cortex. Prenatal ischemia was reported to induce somatosensory map disorganization and neuronal reduction in adulthood 44 . Although we did not detect cell loss in CC projecting neurons, V–VI layer cortical neurons, or corticospinal projection neurons, it is possible that drastic changes in connectivity between areas or cortical map reorganization in motor and somatosensory areas occurred, which could modulate sensorimotor function accompanied with myelination failure. In our preliminary data, cortical map changes following intracerebral microstimulation was shown in the DWMI model brain.
We focused on the neuronal damage responsible for hindlimb sensorimotor function. Several possible reasons are considered in altered dysfunction especially in the hindlimb sensorimotor cortex; further investigations are required to understand the mechanism of altered dysfunction after H-I in the future.
In our preliminary data, the deficit of motor coordination was shown as early as P28 accompanied with myelination failure (Fig. 1E). Similarly, hindlimb motor dysfunction in beam walk ability test and hindlimb retract test was also observed at 2 months (data not shown). Thus, it is noteworthy that a correlation of behavioral impairment with histological damage in myelination was shown in both P26 and P74 (Fig. 1D and E).
Although cystic PVL cases show gray matter damage in some brain areas, including the cerebral cortex, noncystic PVL including DWMI showed less neuronal loss 12 . From our analysis, the DWMI rat model may replicate more diffuse and mild WMI cases without necrosis. This model is useful for pathological analysis of DWMI motor dysfunction induced mainly by myelin deficits without obvious neuronal loss.
Cell-Based Therapies Using the DWMI Model
In the DWMI model that predominantly involves pre-OL degeneration followed by myelination failure, OL lineage cells such as pre-OLs will be an appropriate donor cell type. Embryonic stem cell-differentiated OL lineage cells are suggested to be the preferred cell type for transplantation in the treatment of WMI 26 . The neuroprotective effect of trophic support from grafted OL progenitor cells has recently been reported in a rat PVL model 24 . Therefore, it is very likely that OL lineage cells derived from iPSCs 27 may be an appropriate donor cell type for recovery of motor dysfunction following DWMI.
Regional differences in OL differentiation in the adult brain revealed the role of gray matter and WM niches using homo- and heterotopic transplantation 23 . A preference of O2A progenitors to differentiate into OLs rather than astrocytes was observed in the neonatal rat brain 45 . In this study, cDNA microarray in the P5 sensorimotor cortex revealed 98 upregulated and 65 downregulated genes in the ipsilateral sensory motor cortex compared with the contralateral cortex (Tables 1 and 2). OPC-related genes such as PDGFR-α, NG2, and olig2 were not included in the 163 altered genes, indicating that intrinsic OPCs lost proliferative capacity in gene expression at P5. The gene expression of MBP and PLP myelin protein was not altered by H-I; presumably it is too early to be expressed at this time point. Interestingly, some genes were associated with cell survival, myelin loss, and differentiation of OL lineage cells. Thus, it is possible that replacing lost cells with cells at different stages of maturation (OL lineage cells vs. pre-OLs) has a greater effect than exposure to neurotrophic substances.
Immune responses such as inflammation after H-I and rejection after cell transplantation should be taken into consideration for cell transplantation research. As the neonate has high ability to generate CD4+Foxp3+ regulatory T cells (Tregs) 46 and this ability gradually becomes weaker within 2 weeks after birth, it might be better for us to graft OL lineage cells at earlier stage (P3–P5) after birth when OPCs are proliferating, and the possibility of rejection is less likely. Thus, our DWMI model would be an appropriate model to minimize the rejection after cell transplantation. Future studies should aim to assess OL transplant route, dose, and timing in relation to WMI, OL lineage cell grafts for DWMI, and survival and differentiation of OL lineage cells in vivo. In addition, ways to improve myelination failure of pre-OLs and functional recovery after engraftment need to be addressed.
Footnotes
Acknowledgments
This study was supported by Grants-in-Aid for Scientific Research in priority area (C) (No. 26430020 to H.H.) and young area (B) (No. 23700471, No. 26860851 to S.M.) from the Japan Society for the Promotion of Science (JSPS). H.H. was supported by a grant from the Project for Realization of Regenerative Medicine of the Ministry of Education, Culture, Sports, Science & Technology (MECST). We thank Edanz Group Ltd. for providing language help in editing this manuscript. The authors declare no conflicts of interest.