
Abstract
The use of biomaterials has been demonstrated as a viable strategy to promote cell survival and cardiac repair. However, limitations on combinational cell–biomaterial therapies exist, as cellular behavior is influenced by the microenvironment and physical characteristics of the material. Among the different scaffolds employed for cardiac tissue engineering, a myocardial matrix hydrogel has been shown to promote cardiogenesis in murine cardiac progenitor cells (mCPCs) in vitro. In this study, we investigated the influence of the hydrogel on Sca-1-like human fetal and adult CPCs (fCPCs and aCPCs) when encapsulated in three-dimensional (3D) material in vitro. fCPCs encapsulated in the myocardial matrix showed an increase in the gene expression level of cardiac markers GATA-4 and MLC2v and the vascular marker vascular endothelial growth factor receptor 2 (VEGFR2) after 4 days in culture, and a significant increase in GATA-4 up to 1 week. Increased gene expression levels of Nkx2.5, MEF2c, VEGFR2, and CD31 were also observed when aCPCs were cultured in the matrix compared to collagen. Cell survival was sustained in both hydrogels up to 1 week in culture with the myocardial matrix capable of enhancing the expression of the proliferation marker Ki-67 after 4 days in culture. When encapsulated CPCs were treated with H2O2, an improved survival of the cells cultured in the myocardial matrix was observed. Finally, we evaluated the use of the myocardial matrix as hydrogel for in vivo cell transplantation and demonstrated that the gelation properties of the hydrogel are not influenced by the cells. In summary, we showed that the myocardial matrix hydrogel promotes human CPC cardiogenic potential, proliferation, and survival and is a favorable hydrogel for 3D in vitro culture. Furthermore, we demonstrated the in vivo applicability of the matrix as a potential vehicle for cell transplantation.
Keywords
Introduction
Cardiovascular disease is the leading cause of death in the US with coronary artery disease alone causing one of every seven deaths in 2011 1 . Following a myocardial infarction (MI), cardiomyocyte death is followed by scar deposition and tissue remodeling, causing loss of contractility and impaired cardiac function. At the moment, the only valid option to fully restore the damaged organ is heart transplantation, which is limited by a shortage of donors. For this reason, alternative strategies have been developed to try to regenerate the damaged tissue 2 .
Cellular therapy has emerged as an alternative approach to potentially restore cardiomyocyte loss and cardiac functionality 3 . Different types of stem cells have already been tested in many clinical trials, showing encouraging results in terms of safety, but modest or only transient results in terms of improvement of cardiac function or efficacy4,5. Among the different cell types, cardiac progenitor cells (CPCs) represent a promising cell source for cardiac regeneration for their cardiogenic commitment and capability of differentiating into all of the cell types present in the heart such as cardiomyocytes, endothelial cells, and smooth muscle cells6–10. When transplanted in vivo, human CPCs (hCPCs) have been shown to induce functional regeneration in the infarcted myocardium in small7,9,10 and large11–14 animal models. These encouraging results led to the development of three clinical trials using resident autologous CPCs, resulting in a proof of concept in terms of safety and showing encouraging results in terms of efficacy: CADUCEUS15,16, SCIPIO17,18, and ALCADIA (ClinicalTrials.gov identifier NCT00981006), as well as one clinical trial with heterologous CPCs, ALLSTAR (NCT01458405). However, despite the speed at which stem cells have arrived in the clinic to treat patients with heart disease, the therapeutic mechanism by which they operate remains poorly understood. The idea of using stem cells to repopulate the damaged myocardium was based on the concept that stem cells could differentiate into new cardiomyocytes once the cells are in the cardiac environment because of endogenous stimuli of the damaged cardiac tissue. However, numerous studies have now demonstrated that the extent of cell transdifferentiation in vivo is very limited or absent, and therefore does not explain the observed functional improvements19,20. One proposed mechanism behind the observed therapeutic benefits of stem cells is their paracrine effect, as it has been previously desmonstrated with Akt-modified mesenchymal stem cells (MSCs)21,22. Although committed to the cardiac lineage, CPCs also exert their beneficial therapeutic effects mainly through paracrine signaling, and their contribution to the newly formed cardiomyocytes is only marginal23,24. A possible explanation is the very low cell engraftment25,26 and high cell death due to the hostile environment of the myocardium into which the cells are transplanted 27 .
To overcome these limitations, the use of biomaterials as a carrier has been proposed28–30. Different types of scaffold-based approaches have been shown to increase cell engraftment and survival, either in the form of a hydrogel31–36 or a cardiac patch36–38. However, increasing cell engraftment does not always lead to improved regeneration or an increase of newly formed cardiomyocytes by the transplanted cells 39 . The ideal biomaterial should improve cell engraftment, but it should also promote a proper microenvironment for cell differentiation and protect the transplanted cells from the hostile environment within the infarcted tissue. This should not be surprising, as it is known that stem cell phenotype is influenced by the microenvironment 40 and biomaterial characteristics such as stiffness 41 , biological properties42,43, and architecture 42 .
A myocardial matrix hydrogel derived from decellularized porcine myocardium has recently been developed as an alternative naturally derived biomaterial to treat MI 44 . The main advantage of this material is the presence of tissue-specific extracellular matrix (ECM) components, including proteins and polysaccharides such as glycosaminoglycans 45 . When the myocardial matrix was injected into preclinical MI models, the hydrogel significantly improved global and regional cardiac function45,46, and the success of these studies led to the development of a phase I clinical trial, which is currently ongoing (NCT02305602). Porcine myocardial matrix can also be used as coating material for cell culture 47 , and French et al. showed that the material enhanced cardiogenic commitment of c-Kit+ rat CPCs when compared to collagen-coated dishes 48 . Similar results were also observed when human Sca-1+ CPCs were cultured on coatings of porcineas well as human-derived myocardial matrices 49 . However, the effects of the myocardial matrix on clinically relevant hCPCs cultured in three-dimensional (3D) material still needs to be addressed, as well as the potential of the hydrogel to promote cell survival.
In this study, we investigated the effects of the myocardial matrix on human fetal and adult Sca-1+ CPCs 8 when encapsulated in 3D material in vitro. We also evaluated the ability of the hydrogel to gel with cells in vivo.
Material and Methods
CPC Isolation
Human heart auricle biopsies were obtained under an institutionally approved protocol at the University Medical Center (UMC) Utrecht and the University of California, San Diego Human Research Protections Program.
Human Sca-1+ cells were isolated from human fetal hearts (fCPCs) or from human heart auricle biopsies derived from adult patients (aCPCs), as previously described 8 . The cardiac tissue was cut into small pieces and enzymatically digested in 1 mg/ml collagenase A (Roche, Basel, Switzerland) for 2 h at 37°C, and Sca-1+ cells were isolated by magnetic cell sorting. Cells were cultured in 0.1% gelatin-coated wells in growth medium consisting of 25% endothelial cell growth medium (EGM-2; EGM-2 single quotes; Lonza, Walkersville, MD, USA) in endothelial growth basal medium (EBM-2; Lonza) and 75% M199 (Lonza), 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 1× non-essential amino acids (NEAA; Lonza), and 1× penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA).
Myocardial Matrix Hydrogel Fabrication
Decellularized porcine ventricular ECM was obtained and processed as previously described 50 . Ventricular tissue was isolated from porcine hearts, cut into small pieces, and rinsed in sterile H2O for 2 h. The tissue was then decellularized in 1% sodium dodecyl sulfate (SDS; Thermo Fisher Scientific, San Diego, CA, USA) for 4 days with daily solution changes, rinsed in sterile H2O for an additional day, and frozen at −80°C. Tissue was then lyophilized and milled into a fine powder, which was digested with pepsin (Sigma-Aldrich) at 1 mg/ml in 0.1 M HCl for 48 h at a final concentration of 10 mg/ml. The digested ECM was neutralized to physiological pH, frozen, lyophilized, and stored at −80°C for long-term storage. To reconstitute the matrix, sterile H2O was added to the lyophilized digested matrix at the desired concentration.
Cell Encapsulation
For 3D culture experiments, cells were resuspended in 8 mg/ml myocardial matrix or in 2.5 mg/ml collagen type I (Gibco, Foster City, CA, USA). fCPCs (2 × 105) or aCPCs (1.5 × 105) were encapsulated in 25 μl of myocardial matrix or collagen and incubated at 37°C for 30–45 min to promote hydrogel formation before adding supplemented medium. fCPCs isolated from six different donors and aCPCs from two different donors were respectively pooled together for each experiment. Cells at passages 8 to 12 (after isolation) were used.
Cell Viability Assay
Encapsulated cells were stained with calcein AM viability dye (25 nm; eBioscience, San Diego, CA, USA) for 30 min at 37°C. After incubation, the gels were quickly washed twice in phosphate-buffered saline (PBS; Gibco) and imaged immediately under a Zeiss fluorescence microscope (Carl Zeiss, Dublin, CA, USA) after 1, 4, and 7 days in culture (n = 3).
RNA Isolation and Quantitative Real-Time PCR
Four and 7 days after cell encapsulation, RNA was isolated by NucleoSpin RNAII column (Macherey-Nagel, Mountain View, CA, USA) according to the manufacturer's protocol. DNAse treatment (Macherey-Nagel) was performed on RNA isolation columns to remove DNA contamination. RNA quantification and purity were determined by absorbance readings at 260 and 280 nm using a NanoDrop 2000c spectrophotometer (Thermo Scientific, Wilmington, DE, USA). cDNA was synthesized by using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA), and quantitative real-time polymerase chain reaction (RTq-PCR) amplification was detected in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) using SYBR Select Master Mix (Applied Biosystems, Carlsbad, CA, USA). Four gels at each condition and timepoint were combined and used for RNA isolation. Primers used included the following: GATA binding protein 4 (GATA-4; forward, GTTTTTTCCCCTTTGATTTTTGATC; reverse, AACGACGGCAACAACGATAAT); hNkx2.5 (forward, CCCCTGGATTTTGCATTCAC; reverse, CGTGCGCAAGAACAAACG); myocyte enhancer factor 2c (MEF2c; forward, TCGGGTCTTCCTTCATCAG; reverse, GTTCATCCATAATCCTCGTAATC); myosin light chain 2v (MLC2v; forward, ACCATTCTCAACGCATTC; reverse, CCTAGTCCTTCTCTTCTCC); MLC2a (forward, AACTTCACCGTCTTCCTCAC; reverse, CGAACATCTGCTCCACCTC); α cardiac actin 1 (ACTC1; forward, GTCGGGACCTCACTGACTAC; reverse, CAATTTCACGTTCAGCAGTG); CD31 (forward, GCAGTGGTTATCATCGGAGTG; reverse, TCG TTGTTGGAGTTCAGAAGTG); vascular endothelial growth factor receptor 2 (VEGFR2; forward, AAAGGGTGGAGGTGACTGAG; reverse, CGGTAGAAGCACTTGTAGGC); von Willebrand factor (vWF; forward, AGTGCTGTGATGAGTATGAGTG; reverse, GATGGTGCTTCGGTGGAC); vascular endothelial cadherin (VE-cadherin; forward, GCCAACATCACAGTCAAG; reverse, GCCATATCCTCGCAGAAG); and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH; forward, CTCTGACTTCAACAGCGACA; reverse, TCTCTCTCTTCCTCTTGTGC).
Histological Analysis
For histological analysis, three gels under each condition and timepoint were washed in PBS, embedded in O.C.T. Compound (Sakura Finetek, Torrance, CA, USA), and cut into 7-μm cryosections. Frozen sections were rehydrated in PBS (10 min), fixed in 4% paraformaldehyde (PFA; Thermo Fisher Scientific) (10 min), and washed in PBS (3 × 10 min). Tissue sections were stained with hematoxylin and eosin (H&E; Thermo Fisher Scientific) to evaluate cell distribution and nuclear density within the scaffolds.
For immunofluorescence staining, fixed sections were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich), dissolved in 1% bovine serum albumin (BSA; Gemini Bio Products, West Sacramento, CA, USA) in PBS for 10 min, and blocked with 10% goat serum (Gibco) in PBS for 60 min. The slides were incubated overnight with primary antibody diluted in 0.1% BSA in PBS at 4°C. Secondary antibody incubation was performed at room temperature for 1 h, followed by 5-min Hoechst 33342 (1:5,000) incubation and mounting with fluoromont (Sigma-Aldrich). The following antibodies and dilutions were used: Ki-67 (1:100; Abcam, Cambridge, MA, USA), human β-integrin (1:50; Santa Cruz Biotechnology, Dallas, TX, USA), Nkx2.5 (1:200; Santa Cruz Biotechnology), Alexa Fluor 488 goat anti-mouse (1:200; Invitrogen, Carlsbad, CA, USA), and Alexa Fluor 568 goat anti-rabbit (1:200; Invitrogen). Control slides were incubated with secondary antibody only.
Hydrogen Peroxide Assay
Twenty-four hours after cell encapsulation, gels were treated with growth medium supplemented with 500 μM H2O2 (fCPCs) or 750 μM H2O2 (aCPCs) and incubated for 16 h. After the treatment, the medium was removed, and the gels were incubated in alamarBlue (1:10 in medium; Invitrogen) for a total volume of 250 μl in each well. After 4 h of incubation, 100 μl was collected per sample and analyzed at 550/585 nm with a Synergy 4 Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA) as an index of viable cells. Untreated H2O2 control samples were used to evaluate the percentage of surviving cells in each condition.
In Vivo Application
All animal experiments were carried out in accordance with the guidelines established by the Institutional Animal Care and Use Committee (IACUC) at the University of California in San Diego and the Association for the Assessment and Accreditation of Laboratory Animal Care and the experimental protocol was approved by the UCSD IACUC.
Female Sprague–Dawley rats were used to assess and evaluate the use of the myocardial matrix as a vehicle for cell delivery into the myocardium. Animals were anesthetized using 5% isoflurane (Vet One, Boise, ID, USA), which was lowered to 1–1.5% during surgery. The hearts were exposed via a thoracotomy between the third and fourth ribs, and 3 × 106 fCPCs were resuspended in 75 μl of myocardial matrix (8 mg/ml) and injected into the left ventricular wall. After 30 min, the animals were euthanized, and the hearts were frozen in O.C.T. compound for histological analysis (n = 3).
Statistical Analysis
All data are expressed as the mean ± standard error of the mean (SEM). A paired Student's t-test was used to compare two groups. A value of p < 0.05 was considered significant. Statistical analyses were carried out using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Cell Viability
To assess cell viability and morphology in both myocardial matrix and collagen hydrogels, encapsulated cells were stained with calcein AM vital dye. No major differences were observed between collagen and myocardial matrices or between adult and fetal CPCs. At 1 day, viable cells were homogeneously dispersed within the hydrogel (Fig. 1A), with the majority of them showing a spindle-shaped/elongated phenotype indicative of a good adhesion to the myocardial matrix hydrogel (Fig. 1A). Few cells with round morphology were also present (Fig. 1A). After 4 days in culture, the cells colonized the entire scaffold, indicating proliferation and successful cell migration into the matrix (Fig. 1B). After 7 days in culture, the cells continued to proliferate, showed a very high confluency (Fig. 1C), and also displayed a more elongated phenotype and cell-to-cell contact throughout the gel (Fig. 1D–F).

Representative fluorescent images of calcein AM-labeled human fetal cardiac progenitor cells (CPCs) cultured in myocardial matrix for 1 (A), 4 (B), and 7 (C) days. (D–F) High magnification of (C). Scale bars: 100 μm (A–C), 50 μm (D–F).
RT-PCR Analysis
To evaluate the effects of the myocardial matrix hydrogel on the CPC gene expression profile, quantitative PCR analysis was performed at days 4 and 7 postencapsulation and compared to the collagen hydrogel group.
Fetal CPCs
At day 4, fCPCs showed a significant increase in the early transcription factor GATA-4 (4.4 ± 0.86), the sarcomeric marker MLC2v (6.1 ± 1.6), and the vascular marker VEGFR2 (3.1 ± 1.01) (Fig. 2). Although not significant, increases in the cardiac markers Nkx2.5 (1.9 ± 0.4), MEF2c (2.0 ± 0.52), actin (ACTC1; 4.8 ± 1.69), and MLC2a (2.3 ± 0.73) were observed, while no differences were present in the vascular markers CD31 (1.3 ± 0.13), vWF (1.0 ±0.15), and VE-cadherin expression (VE-CAD; 1.1 ± 0.1) (Fig. 2). Expression of GATA-4 remained significantly increased after 1 week in culture (5.2 ± 1.78), while nonsignificant increases were observed for the other cardiac and vascular markers analyzed: Nkx2.5 (2.1 ± 0.85), MEF2c (2.3 ± 0.99), actin C1 (3.9 ± 1.62), MLC2a (4.3 ± 2.03), MLC2v (2.5 ± 1.44), VEGFR2 (3.2 ± 1.30), CD31 (3.0 ± 1.34), vWF (34 ± 1.63), and VE-CAD (1.4 ± 0.1) (Fig. 2).
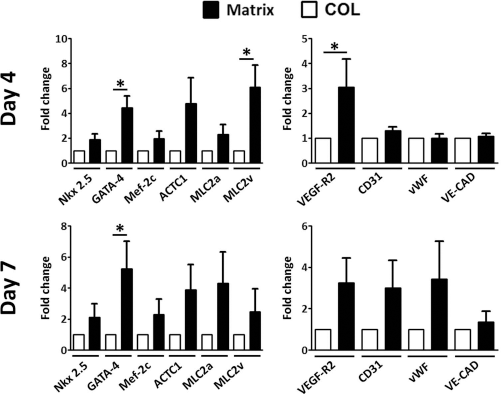
Quantitative polymerase chain reaction (PCR) analysis of fCPCs encapsulated in collagen (COL) or myocardial matrix (Matrix) scaffold. Results are expressed as a fold change for Matrix group over COL; n = 6; *p < 0.05. ACTC1, α cardiac actin 1; MLC2a, myosin light chain 2a; MLC2v, myosin light chain 2v; VEGFR2, vascular endothelial growth factor receptor 2; vWF, von Willebrand factor; VE-CAD, VE-cadherin.
Adult CPCs
Similar to fCPCs, aCPCs also showed an increase in cardiogenic commitment when cultured in the myocardial matrix hydrogel compared to collagen. After 4 days in culture, a significant increase in the early cardiac markers Nkx2.5 (2.4 ± 0.4), MEF2c (1.9 ± 0.33), and the vascular markers VEGFR2 (2.0 ± 0.2) and CD31 (1.9 ± 0.15) was observed (Fig. 3). A nonsignificant increase in GATA-4 was also observed (3.2 ± 0.96), while very low or no expression of sarcomeric markers was detected with no differences among the two groups. After 1 week in culture, no differences were detected among the two groups for the analyzed markers: Nkx2.5 (1.2 ± 0.30), GATA-4 (1.1 ± 0.21), MEF2c (1.1 ± 0.11), VEGFR2 (1.3 ± 0.25), and CD31 (1.9 ± 0.44) (Fig. 3).

Quantitative PCR analysis of aCPCs encapsulated in collagen (COL) or myocardial matrix (Matrix) scaffold. Results are expressed as a fold change for Matrix group over COL; n = 5; *p < 0.05.
Histological Analysis
Histological analysis was performed to evaluate cell phenotype and the proliferation of the cultured cells. H&E staining was performed after 7 days in culture and showed a very high nuclear density for all the groups, confirming that the cells were able to proliferate and colonize the scaffolds (Fig. 4A and B), as previously observed with the cell viability assay (Fig. 1). Expression of Nkx2.5 was also evaluated by immunofluorescence staining. The majority of positive cells were found closer to the gel boarders with few differences among the groups (Fig. 4C and D). We then evaluated the proliferation rate of the cultured cells by Ki-67 immuno fluorescence after 4 days in culture. A significantly higher expression of the proliferation marker Ki-67 was observed both in fetal and adult CPCs when cultured in the myocardial matrix hydrogel. Specifically, 27 ± 3.4% of cells were Ki-67+ when fCPCs were cultured in the matrix, while only 9 ± 1.3% of cells were Ki-67+ when encapsulated in collagen (Fig. 5). A similar trend was also observed with aCPCs, which had 18 ± 3.3% Ki-67+ cells in the matrix compared to 7 ± 0.6% Ki-67+ cells when cultured in collagen (Fig. 5).

Histological analysis of encapsulated fetal CPCs. Hematoxylin and eosin staining of fCPCs (A and B) cultured in myocardial matrix (A) or collagen (B) for 1 week. Immunofluorescence analysis of fCPCs expression of Nkx2.5 (red) and human β1-integrin (green) 4 days after encapsulation in myocardial matrix (C) or collagen (D). Scale bars: 100 μm (A and B) and 50 μm (C and D).

Ki-67 quantification of encapsulated CPCs in collagen (COL) or myocardial matrix (Matrix) 4 days after encapsulation. (A) fCPCs encapsulated in COL or myocardial matrix scaffold; n = 6. (B) aCPCs encapsulated in COL or myocardial matrix scaffold; n = 4; *p ≤ 0.05.
Hydrogen Peroxide Assay
To evaluate whether the myocardial matrix hydrogel has the ability to preserve cell viability compared to collagen, an H2O2 assay was performed. An alamarBlue metabolic assay was used to assess cell survival. A significantly higher metabolic activity was observed when fCPCs (49 ± 5.0 vs. 30.0 ± 1.9%) and aCPCs (75 ± 4.2 vs. 64 ± 2.8%) were encapsulated in the myocardial matrix hydrogel compared to the collagen group (Fig. 6).

H2O2 assay of hCPCs encapsulated in collagen (COL) or myocardial matrix (Matrix) scaffold. (A) Fetal CPCs treated with 500 μM H2O2 for 16 h (n = 12). (B) Adult CPCs treated with 750 μM H2O2 for 16 h (n = 10). Survival was calculated by alamarBlue metabolic activity assay and calculated as percentage versus untreated control samples. *p ≤ 0.05.
In Vivo Transplantation
We also evaluated the ability of the myocardial matrix hydrogel to form a scaffold in vivo when delivering cells into the heart. Histological analysis of cells injected in matrix into the ventricular wall of healthy rats showed that the myocardial matrix hydrogel was still able to form a gel in vivo and was not affected by the presence of the cells. fCPCs were mostly localized in the porcine matrix, and only a few single cells were visible outside of the formed gel (Fig. 7).

H&E staining of transplanted fCPCs in myocardial matrix 30 min after transplantation into the myocardium. Myocardial matrix is outlined by dashed line. Scale bars: 1 mm (A), 100 μm (B); n = 3.
Discussion
Adult stem cell therapy has emerged in the last 15 years as a promising therapeutic approach to repair damaged myocardium. Since the first stem cell transplantation into the infarcted myocardium 51 , many different cell types have been tested both in small and large animal models, as well as in many clinical trials4,5,52. Among the different cell types used, CPCs are very promising cells given their natural commitment to the cardiac lineages and their ability to differentiate in vivo and ex vivo into all of the main cell types present in the heart6,7,9,10. Despite the encouraging results in animal models or clinical trials, limitations such as cell engraftment, survival, and differentiation after transplantation remain a major issue 29 . A cellular therapy that incorporates the proper biomaterial may have the potential to regenerate the damaged tissue, either alone or in combination with bioactive molecules28,53,54. It is now well established that cell transplantation with biomaterials results in an increase in cell engraftment31–38. However, an increase in cell number does not necessarily lead to an increase in regeneration, and many studies have shown that the administration of cells or biomaterials alone has almost the same beneficial effect when the two are administered together 29 . The ideal biomaterial should not only increase cell engraftment but also provide the proper environment for the transplanted stem cells in order to increase their paracrine activity. Most importantly, the biomaterial should also support differentiation of the transplanted cells into newly formed cardiomyocytes, a goal that remains one of the major limitations of current cell therapy applications. Our previous studies demonstrated that CPCs can be cultured in 3D scaffolds and that the properties of the ECM can influence the proliferation rate, metabolic activity, and differentiation potential of the cultured cells37,55,56.
In this study, we evaluated the effects of myocardial matrix-encapsulated hCPCs and showed the ability of this cardiac-derived material to enhance the cardiogenic commitment of both fetal and adult CPCs. fCPCs had increased expression of the early transcription factor GATA-4, the sarcomeric protein MLC2v, and the vascular marker VEGFR2, and although not significant, a general increase of other cardiac markers such as Nkx2.5, MEF2c, actinC1, and MLC2a was observed over the culture period. Similarly, the myocardial matrix also induced a significant increase of Nkx2.5, MEF2c, actin (ACTC1), VEGFR2, and CD31 after 4 days in culture. This effect was more pronounced at this early timepoint after encapsulation. A possible reason is the very high confluency that the cells reach after 1 week in culture together with a possible lack of nutrients that usually limits long-term 3D culture. Another potential explanation is based on the fact that CPCs can remodel their microenvironment. This has been shown in 2D 48 and 3D37,57, and therefore in these experiments, the myocardial matrix may only be providing early cues to promote cardiac differentiation.
Besides the observed cardiogenic commitment, another important aspect of the beneficial effects of the myocardial matrix on CPCs is the increase in proliferation of the cultured cells. We showed that increased numbers of fCPCs and aCPCs were still proliferative in matrices, as shown by the expression of Ki-67, compared to cells in collagen. Promoting proliferation could be an advantage due to the limited number of engrafted cells after transplantation. In addition to an increase in cell retention by injectable scaffolds, an increased proliferative capability of the transplanted cells may also result in an improvement in cardiac regeneration either through an increase in new cardiomyocyte generation or through prolonged paracrine activity.
Cell preservation and survival postinjection are other important aspects, considering the hostile environment of the infarcted myocardium. We showed that the myocardial matrix improved cell survival of encapsulated cells in the presence of H2O2. We observed an approximately 10% increase in cell survival when aCPCs were encapsulated in matrix compared to collagen, and even more when fCPCs were tested. This in vitro result, along with the observed increase in proliferation, shows potential for improved cell survival and proliferation also in vivo. However, further studies are needed to evaluate whether this also occurs in vivo.
One advantage of the myocardial matrix hydrogel is its ability to be delivered through a cardiac injection catheter45,46, therefore allowing a minimally invasive delivery route. Although we did not evaluate this catheter delivery method, we showed that the myocardial matrix can still be injected with cells using a 27-gauge needle and gel in vivo. Our current work demonstrated that in vivo gelation properties of the myocardial matrix were not affected by the presence of the cells. Thirty minutes after injection, cells were visible in the ventricular wall and were located mostly within the formed gel, with few single cells dispersed in the ventricular tissue.
In conclusion, we showed that a naturally derived, cardiac-specific hydrogel enhanced the cardiogenic commitment, proliferation, and survival of 3D-cultured hCPCs when compared to a collagen gel. To our knowledge, this is the first study that evaluated the interaction between tissue-specific ECM and CPCs in 3D. These results provide proof of concept for using a myocardial matrix hydrogel to deliver CPCs in vivo and warrant further investigation in preclinical MI models.
Footnotes
Acknowledgment
This research was supported in part by the National Institutes of Health (NHLBI) through R01HL113468 (to K.L.C.), the Netherlands CardioVascular Research Initiative (CVON), the Dutch Heart Foundation, the Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, the Royal Netherlands Academy of Sciences (to J.P.G.S.), and the Leduq Foundation through a transatlantic career development young investigator award (to R.G.). The authors would like to thank Dr. Mary Nguyen and Jessica Ungerleider for their thoughtful revisions on this article. Dr. Christman is a cofounder, board member, consultant, and holds equity interest in Ventrix Inc.