
Abstract
The aim of this study was to evaluate the overexpression of genes central to cell survival and angiogenesis to enhance the function of human late outgrowth endothelial progenitor cells (EPCs) and their utility for infarct recovery. Ischemic myocardial injury creates a hostile microenvironment, which is characterized by hypoxia, oxidative stress, and inflammation. The infarct microenvironment prevents adhesion, survival, and integration of cell transplants that promote neovascularization. EPCs are dysfunctional as a result of risk factors in cardiovascular patients. Protein kinase B (Akt) and heme-oxygenase-1 (HO-1) are intracellular proteins that play an important role in angiogenesis and cell survival. Late outgrowth EPCs transduced ex vivo with Akt and HO-1 demonstrate improved adhesion to extracellular matrix, improved migration toward human cardiomyocytes, and an improved paracrine profile under stress. Enhanced late outgrowth EPCs reduce the tumor necrosis factor-α (TNF-α) burden both in vitro and in vivo, attenuating nuclear factor-κB (NF-κB) activity and promoting cell survival. Akt and HO-1 enhance late outgrowth EPC neovascularization, resulting in improved cardiac performance and reduced negative remodeling after myocardial infarction in nude mice. Alteration of the infarct microenvironment through gene modification of human late outgrowth EPCs enhances the function and integration of transplanted cells for restoration of cardiac function.
Keywords
Introduction
Myocardial ischemia results in cell death. Inflammation of the ischemic zone is necessary to remove necrotic material, but it also contributes to oxidative and inflammatory stress, which exacerbates secondary necrosis and negative remodeling (39). Revascularization of the ischemic and infarct border zone salvages myocardial tissue and limits stress and negative remodeling (9). Without reperfusion, infarct healing is limited and the risk of heart failure rises. Reparative cells in the myocardium or from the circulation release paracrine factors that promote revascularization and enhance myocardial recovery (9,19). Circulating endothelial progenitor cells (EPCs) (2) are of bone marrow origin and play an important role in reendothelialization of injured vessels and neovascularization of ischemic tissue (9,19). EPCs are dysfunctional in cardiovascular patients compared to healthy subjects, limiting the therapeutic potential of this cell type (9,19). Research into EPCs has also been hindered by the controversy in defining EPC ontogeny, lineage, and physiology (2,4,34). However, strategies aimed at increasing the therapeutic utility of EPCs by genetic means still hold promise for rejuvenating or enhancing EPCs of cardiovascular patients.
Protein kinase B (Akt) is associated with neovascularization and vessel stability (26). Akt loss of function studies result in concurrent loss of EPC motility, reduced nitric oxide bioavailability, and accelerated necrosis in ischemic hind limb models (1). Gnecchi et al. (15) previously used rodent mesenchymal stem cells (MSCs) expressing constitutively active Akt for ischemic injury recovery, revealing the importance of paracrine contributions in cell-based therapy and establishing the paracrine hypothesis. However, the paracrine effects of EPCs are still being elucidated and have been shown to play an important role in wound healing, which includes actions on multiple cell types (10,22,45), and although MSC differentiation to vascular endothelium can occur, it has been an inconsistent finding among researchers (23,28,38).
The stress-induced cytoprotective protein heme-oxygenase-1 (HO-1) also has a role in endothelial protection and function, with particular emphasis on neovascularization recently reviewed by Dulak et al. (12). In HO-1 knock-out mice, EPC tube formation and migration were impaired, in part due to an abnormal response to stromal cell-derived factor-1α (SDF-1α) (11). After hind limb ischemia in rodents, blood flow recovery was impaired in the presence of an HO-1 inhibitor (29), whereas transient overexpression of HO-1 in postischemic tissue promoted recovery and capillary density, partially through stem/progenitor cell recruitment (25,41). HO-1 expression in rodent MSCs was also shown to enhance the benefit of cell therapy (42).
We previously established a codependency between Akt and HO-1 in cellular protection and function in human smooth muscle cells (6). We postulate that the combined expression of these two central regulators of neovascularization and cellular protection could provide human late outgrowth EPCs with an enhanced functional ability in the infarct microenvironment.
Materials and Methods
Human Late Outgrowth EPCs and Cardiomyocytes
Human late outgrowth EPCs were obtained from the mononuclear fraction of whole peripheral blood. The Research Ethics Board of the University Health Network approved the experimental protocol for obtaining and experimenting with human peripheral blood, which was obtained with informed consent. Procedurally, human blood (100 ml) was isolated under sterile conditions by phlebotomy of the cephalic vein and mixed with 100 U heparin (Organon, Toronto, ON, Canada). Mononuclear cells were isolated using Ficoll-Paque™ PLUS (Amersham, Oakville, ON, Canada) according to the manufacturer's instructions and plated onto human fibronectin-coated dishes (BD Biosciences, Bethesda, MA, USA). Cells were incubated for 24 h in endothelial growth factor-supplemented media (EGM-2 bullet kit, Lonza, Basel, Switzerland). EGM-2 was replaced after the first 24 h and every 3 days thereafter. Progenitor cell colonies appeared 15–20 days after initial seeding and were expanded to the third passage in fibronectin-coated dishes. Late outgrowth EPCs were used in the fourth or fifth passage for experimental protocols. Human cardiomyocytes were purchased from a commercial supplier (PromoCell, Heidelberg, Germany) and cultured according to the company's recommendations.
Late outgrowth EPCs were washed once and cultured in minimal basal media (EBM; Lonza) for 24 h to minimize potential confounding growth factor effects from the rich EGM-2 media, then transplanted to the ischemic myocardium in vivo or exposed to a simulated infarct-like environment (IE) in vitro. The in vitro IE was created by incubating late outgrowth EPCs or human cardiomyocytes with human tumor necrosis factor-α (TNF-α) (20 ng/ml; #210-TA, R&D Systems, Minneapolis, MN, USA) (3,17,32,43) and hydrogen peroxide (500 μM; Sigma-Aldrich, St. Louis, MO, USA) (13,18, 21,44) in an external humidified hypoxic environment of 1% O2 and 5% CO2 (8,24,27,40). Glass-bottom delta-T dishes (Bioptechs, Butler, PA, USA) were precoated with 100 μl of Geltrex™ (growth factor-reduced matrix gel; Invitrogen, Burlington, ON, Canada) in EBM. Geltrex™ was added to dishes on ice, leveled with a circular level for 10 min, and incubated at 37°C to solidify on a level surface for 30 min prior to use. Three-dimensional late outgrowth EPC culture (capillary-like tube formation assay) was performed by uniformly seeding late outgrowth EPCs. The duration of IE exposure was as follows: MTT assay, 24 h; nuclear factor κ-light-chainenhancer of activated B cells (NF-κB) analysis, 6 or 24 h; oxidative stress analysis, 6 h; tube formation analysis, 6 h; adhesion assay, 2 h; migration assay, 30 min for lower chamber and 16 h for the upper chamber.
Retroviral Transduction
Late outgrowth EPCs were plated in EGM-2 media at 20–30% confluence. After 24 h, cells were transduced with 10 multiplicities of infection (MOI) of murine stem cell virus (MSCV) expressing HO-1 (HO-1 EPCs), 10 MOI of MSCV expressing Akt (Akt EPCs), or both (Akt/HO-1 EPCs) or, as control, 20 MOI of MSCV expressing green fluorescent protein (EPCs) in EGM-2 media, supplemented with 4 μg/ml of Polybrene® (Sigma) for 36 h, as previously described (7).
Cell Viability
Cell viability was determined by the conversion of MTT [3-(4,5-dimethylthiaxol-2-yl)-2,5 diphenyl tetrazolium bromide] to formazan utilizing nicotinamide adenine dinucleotide (NADH) and NADPH (NADH phosphate) pyridine nucleotide cofactors. MTT was added to a final concentration of 0.5 mg/ml, incubated for 4 h, and solubilized for 24 h at 37°C according to the recommendation of the manufacturer (#11465007001, Roche, Mississauga, ON, Canada). Absorbance was read using a SPECTROMAT® plate reader at 550 nm/690 nm.
Intracellular Redox Environment
Late outgrowth EPCs or Akt/HO-1 EPCs were exposed to an IE for the reported duration or kept at 37°C in 5% CO2/21% O2 for control. Assessment of the intracellular redox imbalance of EPCs was performed using the cell permeable probe CM-H2DCFDA (chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester; 5 μM) preloaded into cells in six-well dishes for 30 min prior to exposure to the IE. Additionally, dihydroethidium (DHE; 1 μM) was added for the last hour of IE treatment. EPCs were washed once with Hanks balanced salt solution (HBSS; Invitrogen), detached with HyQTase™ (Hyclone, Logan, UT, USA), and analyzed by fluorescence activated cell sorting (FACS). General nonspecific oxidation was recorded in the FL1 channel (green) as CM-H2DCFDA was oxidized to dichlorodihydrofluorescein (DCF), while the oxyradicalfavored oxidation of DHE to ethidium was recorded in the FL4 channel (red).
Western Immunoblot
Late outgrowth EPC or Akt/HO-1 EPC lysates were prepared using TPER protein extraction reagent (Pierce, Rockford, IL, USA) containing protease and phosphatase inhibitor cocktails (Sigma). Protein concentration was determined using the Bradford or Lowry method. Cardiac tissue was excised, snap frozen, and pulverized frozen with mortar and pestle. Protein samples (25 μg) were denatured in Laemmli buffer, resolved by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene fluoride (PVDF) membrane (Immobilon-P, 0.45 μm, Millipore, Billierica, MA, USA). Equal loading was verified by Ponceau S, Coomassie stain, and reprobing for β-actin (1:1000; #A5441, Sigma). Membranes were blocked overnight and probed for human HO-1 (1:5000; #SPA-896, StressGen, Victoria, BC, Canada), total Akt and phosphorylated Akt-S473 (1:1000; #9272 and #9271, respectively, Cell Signaling, Boston, MA, USA), total inhibitor κ B (IκBα), and phosphorylated IκBα-pSer32 (1:1000; #PC142 and #400002, respectively, Calbiochem, Gibbstown, NJ, USA), and TNF-α (1: 1000; #SC-1348, Santa Cruz, USA). Blots were then incubated with secondary horse radish peroxidase (HRP)-conjugated anti-rabbit (1:5000; Cell Signaling) or HRP-conjugated anti-mouse (1:5000; Amersham). Immunoreactivity was detected using Chemiglow reagent (Pierce) with film or an Alpha Innotech™ 8900 gel documentation system.
Lentiviral NF-κB Transactivation Promoter-Reporter Assay
Lentivirus Construction
A PmeI/EcoRI/NheI/PstI/ SalI/BspEI/HpaI/BsiwI/EcoRV polylinker (5′-CACCGTTTAAACGAATTCGCTAGCCTGCAGGTCGACTC CGGAGTTAACCGTACGGATATC-3′) was cloned into pLenti6/V5-D-TOPO (Invitrogen; http://tools.invitrogen.com/content/sfs/vectors/plenti6v5dtopo_map.pdf) vector to generate a pLenti-linker according to the manufacturer's directions. A 620-bp EcoRI-NcoI fragment containing the internal ribosome entry site (IRES) from plasmid pIRES2-AcGFP1(Clontech, Mountain View, CA, USA) was ligated into a 4.3-kb EcoRI-XbaI fragment of pIRES (Clontech) along with a 1.7-kb NcoI-XbaI fragment containing the firefly luciferase (Luc) gene from plasmid pGL3 (Promega, Madison, WI, USA) to yield plasmid pIRES-Luc. The gene coding for the Aequorea coerulescens green fluorescent protein (AcGFP1) was amplified from pIRES2-AcGFP1 (Clontech) by PCR using EcoRI-containing primer (5′-ATACCGGAATTCCAACCATG GTGAGCAAGGGC-3′) and SacII-containing primer (5′-CTGTCCCCGCGGTCACTTGTACAGCTCATC-3′). The PCR product was digested with EcoRI-SacII and ligated into EcoRI-SacII digested pIRES-Luc to generate pGFP-IRES-Luc. The 2.9-kb EcoRI-SalI fragment containing cassette GFP-IRES-Luc was ligated with EcoRI-XhoI digested vector pLenti-linker to generate pLenti-GFP-IRES-Luc. Subsequently, a pLentiΔCMV-GFP-IRES-Luc was constructed by replacing a ClaI-SpeI (bases: 1824–2474) fragment of pLenti-GFP-IRES-Luc with ClaI-SpeI PCR amplification products (bases: 2392–2474) to delete the CMV promoter. pLentiΔCMV-NF-κB-GFP-IRES-Luc was then constructed by inserting the NF-κB binding sequences of the human vascular cell adhesion molecule (VCAM) promoter (CTGGGTTTCCCCTTGAAGGGATTTCCCTC) into the vector between SpeI and EcoRI sites upstream of the open reading frame of AcGFP1.
Transactivation Analysis
Late outgrowth EPCs or Akt/HO-1 EPCs were passaged to 24-well dishes and incubated in EGM-2 media with 4 μg of Polybrene® and 5 MOI of lentivirus containing the NF-κB promoter-reporter construct for 24 h. Media was replaced with fresh EGM-2 for 24 h, and cells were washed and incubated for 24 h in EBM. Late outgrowth EPCs or Akt/HO-1 EPCs were exposed to IE or control conditions, then lysed for detection of luciferase activity in a Lumat LB 9507 luminometer using a BrightGLO™ luciferase assay kit from Promega (Madison, WI, USA) according to their instructions.
Adhesion Assay
Late outgrowth EPC adhesion to human extracellular matrix (ECM) was assessed using a commercially available kit (#ECM545, Chemicon, Millipore). Human ECM components (fibronectin, vitronectin, laminin, tenascin, collagen I, II, IV) and bovine serum albumin (control) were precoated in 96-well plates. EPCs were detached and resuspended in EBM; 1 × 105 EPCs or Akt/HO-1 EPCs were seeded into each well and incubated for 2 h either in basal or IE conditions. Nonadherent cells were aspirated, and wells were gently washed three times. Adherent cells were lysed and quantified using CyQuant® GR dye (Invitrogen) at a fluorescent reading of 485ex/520em in a FLUOStar Optima plate reader.
Migration Assay
EPC migration was assessed in accordance with the recommended directions of the supplier (#ECM 510, Chemicon). Either normal EBM media or IE was present in the lower portion of a modified Boyden chamber in the presence of 10% fetal bovine serum (FBS; HyClone, Thermo-Fisher Scientific, Ottawa, ON, Canada) as a nonspecific, universal chemoattractant. Otherwise, 5 × 104 EPCs or Akt/HO-1 EPCs were plated into the lower chamber in EBM and allowed to adhere for 1 h. To induce secretion of infarct-stimulated autocrine/paracrine factors, the cells were then exposed to IE for 30 min, after which 5 × 104 EPCs or Akt/HO-1 EPCs were added to the upper chamber and allowed to migrate through an 8-μm polycarbonate barrier toward the lower chamber overnight (16 h) under nominal or IE conditions. EPCs or Akt/HO-1 EPCs in the upper chamber were then discarded, and cells that migrated to the underside of the filter were detached, lysed, and quantified using CyQuant® GR dye (Invitrogen) at a fluorescent reading of 485ex/520em in a FLUOStar Optima plate reader.
Murine Macrophages
C57BL/6 mice (25–30 g; The Jackson Laboratory, Bar Harbor, MA, USA) were sacrificed by CO2 inhalation. The peritoneal exudates were collected on ice after intraperitoneal injection of 3 × 5 ml complete medium (RPMI-1640, Sigma) with 10% FBS, l-glutamine (HyClone), penicillin, and streptomycin (Invitrogen). The exudates were centrifuged (240 × g, 10 min, 4°C), resuspended in complete medium, counted, and plated (2 × 105 cells/dish) in the center of glass-bottom dishes (MatTek, Ashland, MA, USA). After 2 h at 37°C in a 95% air, 5% CO2 incubator, adhered peritoneal macrophages (MΦs) were washed twice with phosphate-buffered saline (PBS) to remove nonadherent cells, supplemented with fresh complete medium including 1 μg/ml lipopolysaccharide (Sigma), and grown for another 18 h.
Myocardial Infarction (MI), Cell Transplantation, and Cardiac Function
The Animal Care Committee of the University Health Network approved all experimental procedures according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Nude female mice (18–20 g, NU/NU CD1 mice) were purchased from the National Cancer Institute (Bethesda, MD, USA). Nude mice were intubated and ventilated with 2% isoflurane prior to thoracotomy and permanent ligation of the left anterior descending coronary artery, as previously described (37). Pallor and echocardiography confirmed ~35% infarction of the left ventricle. At the time of occlusion, 500,000 genetically modified human male EPCs in a volume of 15 μl or 15 μl of medium control were injected into three sites across the myocardial infarct border zone (defined as the area between pallor and red color areas) with a 27-gauge insulin needle. Five groups were analyzed: an equivalent medium volume without cells (medium group), EPCs (EPC group), Akt EPCs (Akt group), HO-1 EPCs (HO-1 group), and Akt/HO-1 EPCs (Akt/HO-1 group).
Echocardiography at post-MI day 7 and 14 was used to determine heart rate, fractional shortening (FS), left ventricular end-diastolic diameter, and left ventricular end-systolic diameter. Mice were sedated with isoflurane. Heart rates were above 450 bpm, and left parasternal images were taken in the right lateral decubitus position using a 13-MHz transducer (Sequioa C256 and 15L8, Acuson, Mountain View, CA, USA). Images were stored as short-axis, two-dimensional, digital loops and M-mode images at the midpapillary level of the left ventricle. At 14 days, pressure–volume analysis was carried out under positive pressure ventilation using a micromanometer conductance 1.4-Fr catheter (Millar Instruments, Houston, TX, USA). Pressure–volume loops were determined during a transient apnea. Preload recruitable stroke work (PRSW) and end-systolic pressure–volume relationship (ESPVR) loops were measured during vena cava occlusion. Load-dependent analysis was used to determine ejection fraction and positive developed pressure over time (+dP/dt).
Immunohistochemistry and Image Analysis
For late outgrowth EPC retention analysis, paraffin-embedded (5 μm thick) sections were incubated with primary antibody specific to human mitochondria (1: 100; #MAB1273, Millipore/Chemicon International). Total late outgrowth EPC retention was assessed by measuring the number of positively stained cells per microscopic field (200x equal to 0.4 mm2) in five fields per slide.
For apoptosis analysis, terminal dUTP nick-end labeling (TUNEL) assay was performed on tissue sections with an in situ Cell Death Detection kit (Roche) according to the instructions of the manufacturer. Apoptotic cell death was determined by counting the number of TUNEL-positive nuclei per microscopic field (200x equal to 0.4 mm2) in five fields per slide.
For angiogenesis analysis, paraffin-embedded (5 μm thick) sections were incubated with primary antibody specific to von Willebrand factor (1:100; vWF-FVIII, Invitrogen). The density of blood vessels was assessed by measuring the number of positive signals per microscopic field (200x equal to 0.4 mm2) in five fields per slide.
For oxidative stress analysis, paraffin-embedded (5 μm thick) sections were incubated with primary antibody specific to the oxidized epitope (8-OH deoxyguanosine) and not the 2′-deoxyguanosine (1:50; #ab26842, Abcam, Cambridge, MA, USA). The area of oxidative stress was assessed by positive signals per microscopic field (200x equal to 0.4 mm2) in five fields per slide.
For scar analysis, the heart (after functional analysis) was formalin-fixed with diastolic pressure at 20 mmHg. After 48-h fixation, the heart was sectioned in 1-mm-thick slices (5–6 sections). We measured all five heart sections from the apex side to calculate the scar area. Specifically, both apical and basal sections were digitally photographed (Coolpix, Nikon, Tokyo, Japan). ImageJ Software was used for morphometric analysis. The epicardial surface areas of the left ventricular free wall (LVFW) and any scar tissue in the LVFW were measured by planimetry. The surface areas of the epicardial scar tissue and the LVFW were measured as the sum of the epicardial length times the section thickness (1 mm). The surface area percentage of scar tissue in the LVFW was calculated as follows: (epicardial scar area)/(epicardial LVFW area) x 100.
Phagocytosis Assessment
Peritoneal MΦs or late outgrowth EPCs were grown for 1 h in minimal medium. India ink (stock solution in PBS; #3398, Speedball, Kingston, ON, Canada) was added in a 1:4000 dilution to the medium. Peritoneal MΦs or late outgrowth EPCs were incubated for 60 min at 37°C or 4°C (for nonspecific binding control), washed four times with HBSS, and assessed by phase contrast microscopy. Yellow–green FluoSpheres (505/515 nm, 0.2 μm diameter; Invitrogen) were washed once in PBS (16,000 × g, 10 min, 4°C), resuspended, and sonicated to dissolve aggregates and used either directly or with prior opsonization using 5% normal goat serum (Jackson ImmunoResearch, West Grove, PA, USA) in PBS for 16 h at 4°C. Peritoneal MΦs or late outgrowth EPCs were incubated for 1 h in minimal medium, and beads were added in a ratio of 1000 beads/cell. After 60 min at 37°C or 4°C (control), cells were washed four times with HBSS, detached using HyQTase™ solution (20 min, 37°C; HyClone), and pelleted by centrifugation. Peritoneal MΦs or late outgrowth EPCs were resuspended in HBSS and analyzed by FACS (FC500, Beckman Coulter, Mississauga, ON, Canada) flow cytometry. The mean fluorescence intensity of cells was compared to control cells previously incubated at 4°C with or without beads, and no difference in EPC groups confirmed the absence of phagocytosis (data not shown).
Late Outgrowth EPC Immunophenotyping/Antigenicity by FACS Flow Cytometry
Late outgrowth EPCs near 80% confluence were detached with HyQTase™ (Hyclone) and washed once with PBS-5% FBS. Late outgrowth EPCs were resuspended in 90 μl PBS-5% FBS and 10 μl of monoclonal-conjugated antibody or isotype control. We used the following antibodies with their respective manufacturer-recommended isotype controls to immunolabel late outgrowth EPCs prior to transduction: CD31-fluorescein isothiocyanate (FITC; #BD-555445), CD34-FITC (#BD-555821), CD54-phycoerythrin (PE; #BD-555511), CD106-FITC (#BD-551146), CD184-allophycocyanin (APC; #BD-555976), 140b-PE (#BD-558821), all from BD Biosciences; CD105-PE (#FAB10971P), CD117-PE (#FAB332P), CD202b-APC (#FAB3131A), CD144-PE (#FAB9381P), CD309-PE (#FAB357P), all from R&D Systems; CD133 (#130-090-826, Miltenyi Biotec, Auburn, CA, USA); and combined CD14-PE/CD45-FITC Multimix (Dako, Mississauga, ON, Canada). Late outgrowth EPCs were placed in the dark on ice with shaking for 1 h. Late outgrowth EPCs were washed twice with PBS–5% FBS and examined by FACS. The percentage of positive late outgrowth EPCs was calculated using the conjugate isotype negative controls according to the method of Overton (31).
Secretome Protein Array
Four independent transductions of late outgrowth EPCs were performed in T25 flasks; each flask was split into three wells of a six-well plate. After exposure to the IE for 24 h, media were collected, pooled, and concentrated using an Amicon Ultra-15, 3 kDa concentrator (#EFC-900308, Millipore) by centrifugation at 4000 × g for 60 min at 4°C. EPC or Akt/HO-1 EPC lysates were collected by adding 50 μl/well of PBS with protease and phosphatase inhibitor cocktails (Sigma), 0.5% Triton X-100, and 0.5% NP-40 detergents. Lysates were collected and homogenized with a plastic Eppendorf pestle, and centrifuged for 5 min at 10,000 × g at 4°C. Conditioned media (undiluted) and supernatant (200 μg/ml) were snap frozen and shipped frozen to RayBiotech, Inc. (Norcross, GA, USA) for proteome profiling using a human G-series 2000 array. Samples were internally normalized and data were presented as integrated density values (IDVs). We used a weighted measure to assign priority to results based on total detectability, arithmetic difference, and relative difference in comparison to parallel control.
Statistical Analysis
All values are expressed as mean ± SD. Comparisons of two parameters used unpaired t-test and parameters among 3–5 groups were analyzed by one-way ANOVA for single-factor or two-way ANOVA for two-factor variables with repeated measures, followed by Newman-Keuls or Bonferroni multiple comparisons. Differences were considered statistically significant at a value of p < 0.05.
Results
Akt/HO-1 EPCs Survive in a High-Stress In Vitro Environment
Human late outgrowth EPCs were characterized for antigens and endothelial function (Fig. 1) and were highly amenable to viral-mediated gene expression (Fig. 2A–C). Transduction efficiency was determined by green fluorescent protein (GFP) analysis (Fig. 2A). Western blot analysis of Akt/HO-1 EPCs demonstrated a significant fourfold elevation in constitutively active Akt and twofold increase in HO-1 expression (Fig. 2B, C). To determine whether Akt/HO-1 EPCs could improve functional recovery, we first assessed their capacity to resist a high-stress “infarct-like” microenvironment (IE) in vitro. Late outgrowth EPCs were exposed to the upper range of physiological levels of human TNF-α (20 ng/ml) and H2O2 (500 μM) in a hypoxic (1% O2) atmosphere. This hostile microenvironment was used to simulate the combined inflammation, oxidative stress, and hypoxia that EPCs would be exposed to after clinical transplantation. Mitochondrial reductase activity was determined by quantification of tetrazole conversion to formazan by viable late outgrowth EPCs at 80% confluence after exposure to an in vitro IE (MTT assay). The viability of late outgrowth EPCs was reduced to <50% as determined by MTT assay (Fig. 2D). In contrast, mitochondrial reductase activity was preserved (>70%) in Akt/HO-1 EPCs. Further, there was a significant reduction in the transactivation of the redox- and inflammatory-sensitive transcription factor NF-κB in Akt/HO-1 EPCs (Fig. 2E, i). This was also accompanied by rapid phosphorylation and subsequent degradation of the IκB protein (Fig. 2E, ii). We also measured a marked reduction in the amount of intracellular oxidative stress in Akt/HO-1 EPCs as determined by the attenuated intensity of the redox-sensitive fluorochromes CM-H2DCFDA and DHE (Fig. 2F). In addition, levels of prosurvival hepatocyte growth factor (HGF), interleukin-8 (IL-8), and osteoprotegerin were elevated in the medium as determined by the Raybiotech proteome array (Table 1). Surprisingly, TNF-α in the medium 24 h after exposure to the IE was reduced by ~50% in Akt/HO-1 EPCs (Table 2). Taken together, these data demonstrate that a pleiotropic benefit is conferred by Akt and HO-1 expression in EPCs that includes modification of paracrine factors of the secretome.
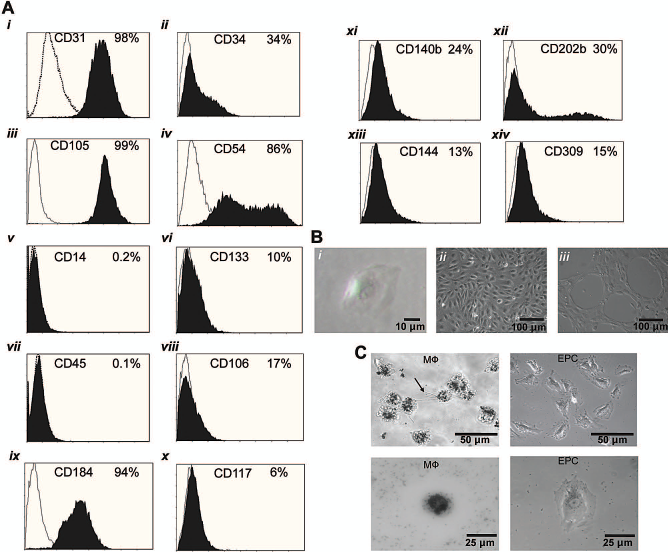
Postisolate characterization of human late outgrowth endothelial progenitor cells (EPCs). (A) Cluster of differentiation (CD) profile of late outgrowth EPCs prior to transduction. Percent positive cells (black histograms) was calculated by the Overton method (31) with their respective matched IgG isotype/fluorochrome controls (white histograms). Antigens and common names: (i) CD31: platelet endothelial cell adhesion molecule (PECAM). (ii) CD34: hematopoietic progenitor cell antigen. (iii) CD105: endoglin. (iv) CD54: intercellular adhesion molecule. (v) CD14: lipopolysaccharide receptor. (vi) CD133: prominin-1. (vii) CD45: leukocyte common antigen. (viii) CD106: vascular cell adhesion molecule. (ix) CD184: fusin. (x) CD117: c-kit receptor. (xi) CD140b: platelet-derived growth factor receptor. (xii) CD202b: tunica interna endothelial cell kinase (TEK). (xii) CD144: vascular epithelium cadherin (VE-cadherin). (xiv) CD309: vascular endothelial growth factor receptor-2 (VEGFR-2). (B) Phase contrast images of isolated EPCs. (i) Single colony-forming late outgrowth EPC. (ii) Confluent monolayer of late outgrowth EPCs. (iii) Unstimulated capillary-like tube formation in growth factor-reduced extracellular matrix (ECM; GelTrex™) in basal media. (C) Phase contrast images of macrophages (MΦs) undergoing phagocytosis of carbon particles (left panels, arrow indicates pseudopod). No phagocytosis is observed in late outgrowth EPCs (right panels). Representative of three experiments.

Improved survival and function of late outgrowth endothelial progenitor cells (EPCs) expressing Akt/HO-1 in a simulated infarct-like environment (IE). (A) Transduction efficiency of human EPCs. Flow cytometric assessment of EPCs transduced with 20 multiplicities of infection (MOI) of murine stem cell virus (MSCV) encoding green fluorescent protein (GFP). Inset panels are phase and fluorescent images in identical fields. (B, C) Western blot analysis of late outgrowth EPCs transduced with MSCV encoding Akt and heme oxygenase-1 (HO-1) showing expression levels of Akt and HO-1 after transduction compared to control (096368912X653002p < 0.0001). (D) Akt/HO-1 EPCs demonstrated improved survival after a 24-h IE exposure by MTT viability assay compared to control [one-way ANOVA; *p < 0.05 vs. normal cell conditions, #p < 0.05 vs. control transduction in IE (n = 10)]. (E, i) Nuclear promoter-reporter assay using nuclear factor κB (NF-κB) repeats upstream of luciferase demonstrated reduced NF-κB transactivation 6 and 24 h after exposure to IE in Akt/HO-1 EPCs [one-way ANOVA; n.s., not significant; *p < 0.05 vs. control (n = 12)]. (ii) Western blot for inhibitor of κB (IκB) is rapidly phosphorylated by 6 h and degraded by 24 h. (F) Oxidative stress fluorophores chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA; green) and dihydroethidium (DHE; red) in control EPCs (top panels) compared to Akt/HO-1 EPCs (bottom panels) after 6-h IE exposure. (i vs. v) Fluorescence in establishing vascular networks in GelTrex™. Flow cytometry for (iii vs. vii) CM-H2DCFDA and (iv vs. viii) DHE fluorescent intensity.
Elevated Protein Expression in Conditioned Medium of Human Akt/HO-1 EPCs Relative to Control

Protein levels were measured 24 h after exposure of endothelial progenitor cells (EPCs) to an infarct-like environment (IE) using the Raybiotech Human G Series 2000 array. Data represent four replicate transductions. Each replicate was passaged in triplicate to six-well culture dishes 24 h prior to IE exposure. Samples were pooled, and post hoc positive control normalization and background subtraction were utilized to determine the integrated density value (IDV) of 174 proteins; values of a weighted sum >3 are reported. HO-1, heme oxygenase-1.
Reduced Protein Expression in Conditioned Medium of Human Akt/HO-1 EPCs Relative to Control

Protein levels were measured 24 h after exposure of endothelial progenitor cells (EPCs) to an infarct-like environment (IE) using the Raybiotech Human G Series 2000 array. Data represent four replicate transductions. Each replicate was passaged in triplicate to six-well culture dishes 24 h prior to IE exposure. Samples were pooled, and post hoc positive control normalization and background subtraction were utilized to determine the integrated density value (IDV) of 174 proteins; values of a weighted sum >3 are reported.
Adhesion and Migration of Akt/HO-1 EPCs Are Augmented
The ability of late outgrowth EPCs to form tubes in Matrigel (Fig. 3A, i) was almost completely lost after exposure to the stress of the IE (Fig. 3A, ii). However, the formation of capillary-like tubes under stress was preserved in Akt/HO-1 EPCs (Fig. 3A, iii). Considering the evidence for reducing free radicals and the toxic cytokine TNF-α, it was not clear whether improved tube formation was simply a consequence of cell survival. Therefore, since adhesion to extracellular matrix (ECM) is an important function of cells forming neovessels and is necessary for juxtacrine signaling to native vessels for angiogenesis after transplantation, we tested whether this was altered in Akt/HO-1 EPCs. Under stress-free conditions, adhesion to ECM proteins, with the exception of collagen I, was significantly improved in Akt/HO-1 EPCs (data not shown). Importantly, the capacity of late outgrowth EPCs to adhere to ECM proteins was impaired by the IE conditions (Fig. 3B). In contrast to observations under basal conditions, Akt/HO-1 EPCs were significantly more adherent to collagen I in the IE. Further, these Akt/HO-1 EPCs elicited greater adherence to some (vitronectin, tenascin, laminin, and collagen II) but not all (fibronectin and collagen IV) ECM proteins in the IE. In part, this could be due to an elevation in the adhesion molecules activated leukocyte cell adhesion molecule (ALCAM), platelet endothelial cell adhesion molecule (PECAM1), and intercellular adhesion molecule (ICAM2) (Table 1) or to the moderate 17% increase in E-selectin measured in cell lysates from Akt/HO-1 transduced EPCs (data not shown).

Improved adhesion and migration of late outgrowth endothelial progenitor cells (EPCs) expressing Akt/HO-1. (A, i) EPCs spontaneously form capillary-like tubes in growth factor-reduced ECM (GelTrex™). The capacity for EPCs to form tubes is lost in the infarct-like environment (IE) (ii) but preserved in Akt/HO-1 EPCs (iii) (representative of three experiments; sampled after 6 h of IE incubation). (B) Akt/HO-1 EPCs in the IE had a significantly higher capacity to adhere to human vitronectin, tenascin, laminin, and collagen I and II after 2 h of IE incubation [one-way ANOVA; n.s., not significant; *p < 0.05 vs. control (n = 6)]. (C) Modified Boyden chamber migration assay with 10% serum, cardiomyocytes, EPCs (for control), and Akt/HO-1 EPCs (for Akt/HO-1) serving as the stimulus for late outgrowth EPC migration. In the IE (30 min for lower chamber and 16 h for upper chamber), Akt/HO-1 EPCs were significantly more motile toward cardiomyocytes and Akt/HO-1 EPCs [n.s., not significant; *p < 0.05 vs. control; average ± SD (n = 6)].
Cellular migration is also an important component of neovessel formation. Under basal conditions, Akt/HO-1 EPCs had no advantage over EPCs in their ability to migrate toward a general stimulus of 10% serum (Fig. 3C). Not surprisingly, the migration of EPCs was severely reduced by ~70% with a stimulus of 10% serum with IE (Fig. 3C). Unexpectedly, Akt/HO-1 EPCs gained no advantage in migration toward serum in the IE (Fig. 3C). However, multiple factors are reportedly responsible for migration or chemoattraction of EPCs to the infarct. Indeed, it is generally accepted that cardiomyocytes under ischemic stress or after reperfusion injury release chemotactic factors to mobilize resident and peripheral circulating EPCs. To better simulate a genuine secretome of cardiomyocytes under stress, we used human cardiomyocytes exposed to the IE without serum as the sole stimulus for late outgrowth EPC migration. The presence of human cardiomyocytes exposed to the IE stimulated migration of EPCs (Fig. 3C). This chemotactic effect was significantly augmented for Akt/HO-1 EPCs (Fig. 3C). Further, EPCs drawn to sites of ischemic injury from the peripheral blood or after transplantation must communicate with each other through chemotactic stimuli to align for neovessel formation. As such, we also examined the effect of EPCs in an IE as a source of paracrine factors that could promote migration. Those Akt/HO-1 EPCs demonstrated significantly greater migration to other Akt/HO-1 EPCs in an IE (Fig. 3C). These data suggest that the collective presence of postinfarct cardiomyocytes and Akt/HO-1 EPCs could potentially attract additional cells through autocrine/paracrine signaling (see Table 1). Taken together, improved adhesion, survival, migration, and an altered secreted protein profile in Akt/HO-1 EPCs could result in greater cell integration at the site of injury after MI. To test this proof-of-principle, we examined the cells in a nude mouse model of MI.
Transplantation of Akt/HO-1 EPCs After MI Improves Cardiac Recovery in Mice
Akt/HO-1 EPCs Show Improved Survival and Neovessel Formation
Using anti-human mitochondria antibodies, we identified the number of human late outgrowth EPCs within the peri-infarct area of the mouse heart 14 days after implantation. The cells were identified morphologically as integrating into small vessels of the heart (Fig. 4A). The number of Akt/HO-1 EPCs was highest (Fig. 4B, C) compared with all control groups (Fig. 4A, C), though the Akt EPC and HO-1 EPC groups did demonstrate higher cellular survival than the EPC control (Fig. 4C). Blood vessel density overall was elevated in the nude mice hearts in the Akt/HO-1 EPC transplant group to a greater extent than with either gene alone (Fig. 4D, E). All late outgrowth EPC groups revealed greater blood vessel density compared with the medium control group.
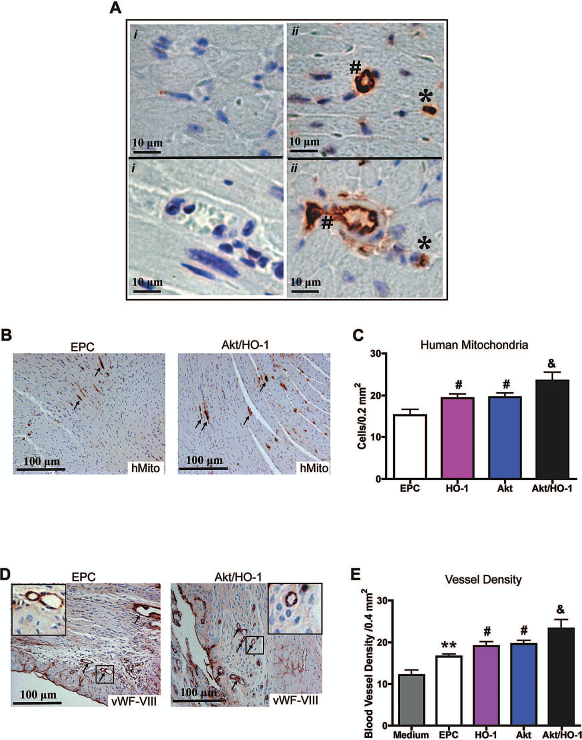
Enhanced cell survival in post-myocardial infarction mouse hearts receiving human late outgrowth endothelial progenitor cells (EPCs) expressing Akt/HO-1 improves host neovascularization. (A, B) Human mitochondrial protein labeled by immunohistochemistry. Human late outgrowth EPCs integrated into small microvessels (ii, #) and capillaries (ii, *); vessels of similar size in distal sections (i) are negative for human-specific mitochondrial protein to demonstrate antibody specificity. (B) Arrows indicate intense staining in microvasculature. (C) Labeling was quantified by planimetry at day 14. (D) Blood vessel density was measured by von Willebrand factor/factor VIII (vWF-FVIII) immunohistochemistry (arrows indicate intense staining in microvasculature; inset magnified 9x) and (E) quantified by planimetry [one-way ANOVA; **p < 0.001 vs. medium control (n = 15); #p < 0.05 vs. EPC control (n = 4); &p < 0.05 vs. Akt EPC control (n = 7) or HO-1 EPC control (n = 5); Akt/HO-1 EPC group (n = 6); Original magnification: 400x (A), 100x (B, D).
Apoptotic cells in the infarct and border region were identified 14 days after implantation (Fig. 5A). The number of TUNEL-positive nuclei was significantly higher in mice treated with medium compared to mice treated with EPCs, Akt EPCs, or HO-1 EPCs (Fig. 5B).
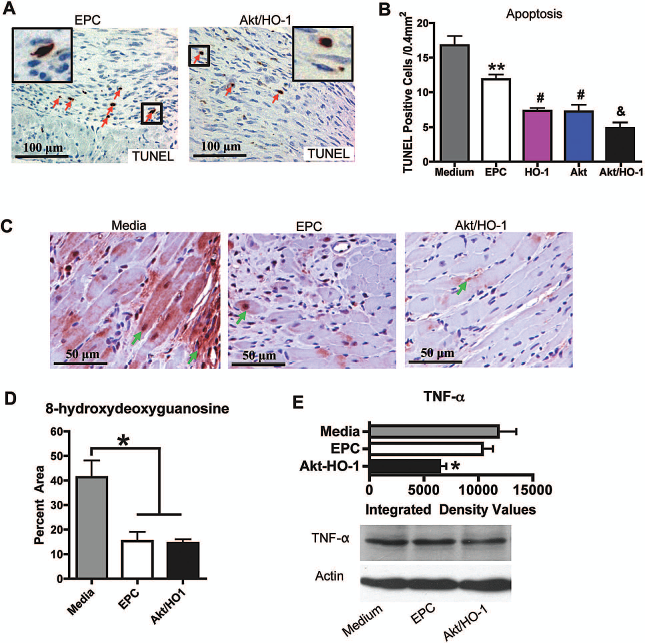
Enhanced cell survival in post-myocardial infarction (MI) mouse hearts receiving human late outgrowth endothelial progenitor cells (EPCs) expressing Akt/HO-1 reduces oxidative stress and tumor necrosis factor-α (TNF-α). (A) TUNEL staining of apoptotic cells (arrows; original magnification 200x inset magnified 9x). (B) Quantification of apoptosis [one-way ANOVA; **p < 0.001 vs. media control (n = 15); #p < 0.05 vs. EPC control (n = 4); &p < 0.05 vs. Akt EPC control (n = 7) or HO-1 EPC control (n = 5); Akt/HO-1 EPC group (n = 6)]. (C) Oxidative stress was measured by 8-hydroxydeoxyguanosine immunohistochemistry (original magnification 400x; arrows indicate intense nuclear staining) and (D) quantified by planimetry (n = 3 per group). (E) Western blot shows Akt/HO-1 group demonstrated significantly reduced TNF-α-immunoreactive protein at day 3 post-MI (n = 3 per group).
TUNEL-positive nuclei were significantly fewer in the Akt/HO-1 EPC group than either the Akt EPC or HO-1 EPC group. Apoptosis at 2 weeks was predominately in host tissue in the peri-infarct region and was contained by the transplantation of Akt/HO-1 EPCs.
To identify the mechanism for superior survival of host cells by Akt/HO-1 EPCs compared to EPCs, we examined the peri-infarct region for oxidative stress. Immunolabeling identified the oxidized derivative of deoxyguanosine, 8-hydroxydeoxyguanosine, that accumulates in the nuclear and mitochondrial DNA of cells with redox imbalance. The degree of oxidative stress in the peri-infarct region was significantly reduced in the hearts receiving EPCs and those receiving Akt/HO-1 EPCs compared to media control (Fig. 5C). The reduction in oxidative stress was not different between EPCs and Akt/HO-1 EPCs. This suggests that late outgrowth EPC transplantation reduces the accumulation of free radical stress, but is unlikely the reason for greater benefit achieved by EPCs expressing Akt/HO-1. However, early experiments 3 days post-MI revealed a significant decline in the amount of TNF-α-immunoreactive protein within the ischemic area where Akt/HO-1 EPCs were implanted (Fig. 5D). This 50% reduction in TNF-α was also observed in our in vitro analysis with high levels of TNF-α (Table 2) added to the medium as a component of the IE. TNF-α is reduced in part through the secretion of soluble TNF receptors. The amounts of soluble TNF receptor 1A/B and 6 were elevated in Akt/HO-1 EPCs in vitro (Table 1). Taken together, EPCs effectively buffer the redox imbalance of post-MI hearts, but the added benefit achieved through Akt/HO-1 gene expression is a reduction in TNF-α. To determine whether these molecular effects translated to functional improvement, we next detailed functional cardiac recovery.
Akt/HO-1 EPCs Confer Improved Cardiac Function After MI
A progressive alteration of cardiac function following cell transplantation was determined using echocardiography. Coronary artery ligation resulted in a significant decrease in left ventricular (LV) function and increase in dilatation after MI. At 7 days post-MI, the transplantation of Akt/HO-1 EPCs demonstrated higher fractional shortening (FS) compared with Akt EPCs or HO-1 EPCs (Fig. 6A). The functional preservation was better in the Akt EPC group than in the EPC group. No difference was observed at day 7 between the HO-1 EPC group and the EPC group, and neither was significantly improved by comparison to medium alone. By day 14 post-MI, the Akt EPC and HO-1 EPC groups demonstrated better cardiac function with higher FS than the EPC group, which also demonstrated improved function compared with the medium control group. However, the Akt/HO-1 EPC group maintained cardiac functional recovery with significantly higher FS than all controls (Fig. 6A).

Cardiac function in post-myocardial infarction (MI) mice shows enhanced recovery with human late outgrowth endothelial progenitor cells (EPCs) expressing Akt/HO-1. (A) Echocardiographic assessment of percent fractional shortening at 7 and 14 days post-MI demonstrates progressive cardiac recovery. Ventricular volumes and cardiac function were evaluated by under load-dependent (B–D) and load-independent (E–G) conditions 14 days post-MI. The Akt/HO-1 EPC group shows synergistic functional improvements, except for end-diastolic volume. (B) Ejection fraction. (C) End-systolic volume. (D) End-diastolic volume. (E) Representative pressure–volume loops. (F) End-systolic pressure–volume relationship (ESPVR). (G) Preload recruitable stroke work (PRSW) [two-way ANOVA; *p < 0.05; **p < 0.001 vs. medium control (n = 15); #p < 0.05, ##p < 0.001 vs. EPC control (n = 4); &p < 0.05 vs. Akt EPC control (n = 7) or HO-1 EPC control (n = 5); Akt/HO-1 EPC group (n = 6)].
On day 14 after MI, cardiac function was evaluated using a pressure–volume catheter under load-dependent and load-independent conditions. Compared to medium and EPCs, Akt EPC, HO-1 EPC, and Akt/HO-1 EPC groups improved load-dependent parameters, including ejection fraction (Fig. 6B) and end-systolic/end-diastolic volumes (Fig. 6C, D), as well as load-independent parameters, including end-systolic elastance (ESP/ESV slope, ESPVR) (Fig. 6F) and preload recruitable stroke work (PRSW) (Fig. 6G). Importantly, the Akt/HO-1 EPC group significantly improved these parameters compared with either the Akt EPC or HO-1 EPC group, with the exception of end-diastolic volume. Akt/HO-1 EPC, Akt EPC, HO-1 EPC, and EPC treatments did reduce end-diastolic volume relative to medium, but no synergistic benefits were found for this parameter (Fig. 6D).
Akt/HO-1 EPCs Reduce Myocardial Scarring and Wall Thinning
Myocardial scar tissue was analyzed (Fig. 7A–E), and the scar, border region, and normal myocardial tissue were identified with Masson's trichrome staining (Fig. 7F–J). Quantitative planimetry revealed more extensive (Fig. 7K) and thinner (Fig. 7L) scars in medium control animals than in those receiving EPCs, Akt EPCs, or HO-1 EPCs. Notably, scarring was less extensive and thicker in animals that received the Akt/HO-1 EPCs (Fig. 7J) in comparison to Akt EPCs or HO-1 EPCs (Fig. 7H, I), which suggests that the combination of Akt and HO-1 enhanced the capability of late outgrowth EPCs to stabilize the infarct and prevent scar expansion and ventricular dilatation, in agreement with functional parameters.

Morphological analysis of post-myocardial infarction mouse hearts shows improved remodeling with human late outgrowth endothelial progenitor cells (EPCs) expressing Akt/HO-1. (A–E) Myocardial scar tissue. Arrows indicate heart apex; dashed lines indicate infarct area. (F–J) Whole heart sections stained with Masson's trichrome. (K) Quantitative analysis of scar area and (L) thickness [one-way ANOVA; *p < 0.05; **p < 0.001 vs. medium control (n = 15); #p < 0.05 vs. EPC control (n = 4); &p < 0.05 vs. Akt EPC control (n = 7) or HO-1 EPC control (n = 5); Akt/HO-1 EPC group (n = 6)].
Discussion
This study demonstrates that late outgrowth EPCs expressing Akt and HO-1 are better able to prevent cardiac dysfunction after an MI. Late outgrowth EPCs expressing Akt and HO-1 are better able to adhere, survive, and integrate functionally after MI. Transplantation of late outgrowth EPCs to the postinfarct environment first requires adhesion to the ECM for cell survival and integration. Following cell adhesion, late outgrowth EPCs migrate to form neovessels and communicate through paracrine signaling to promote host angiogenesis. Our study directly targeted upstream intracellular mechanisms important for late outgrowth EPC function as a strategy to enhance the therapeutic potential of late outgrowth EPCs.
Expression of Akt/HO-1 attenuates intracellular oxidative stress and the proinflammatory transcription factor NF-κB in late outgrowth EPCs. This could, at least in part, be a component of improved function in the infarct environment. Naturally, NF-κB is likely not the sole regulated pathway, though hypoxia acts through Akt-mediated NF-κB to regulate neovascularization (35). For example, TNF-α can also activate hypoxia inducible factor 1α (HIF1-α) in an NF-κ-dependent manner, even under normoxic conditions (20,46). Early post-MI activation of NF-κB is heavily influenced by inflammation and the levels of TNF-α released by resident mast cells and other infiltrating leukocytes. TNF-α negatively influences EPC function, neovascularization, and myocardial recovery after infarction (16). This study provides a mechanism for reducing cardiac dysfunction with Akt/HO-1 EPCs through reduced oxidative stress in addition to the elimination of TNF-α by ~50% both in vitro and in vivo. Though at low relative abundance, there was evidence of soluble TNF receptors 1A/B and 6 being elevated in the medium of Akt/HO-1 EPCs in vitro. As such, other mechanisms may be involved in the reduction of TNF-α. As a limitation of our study, we chose TNF-α to represent the stress of inflammation for its cytotoxic effects in the post-MI environment and have not examined other cytokines (14,17). However, regulating TNF-α through cells capable of adapting to the local microenvironment could provide a better option than direct TNF-α targeting, which has so far produced disappointing clinical outcomes (i.e., RENEWAL) (29). The use of TNF-α antibodies has previously been shown to be effective in preclinical models (5). However, achieving the appropriate localized effects is a challenge to clinical antibody therapy, and TNF-α has receptorand dose-dependent physiological roles (36). A cell-based therapeutic approach allows for adaptation of the local microenvironment that is unavailable with antibody therapy. In this way, transplanted cells could protect against pathophysiological levels of TNF-α without abrogating its physiological purposes and could thus offer a superior alternative. To our knowledge, this is the first evidence that cell therapy is capable of providing a “molecular sink” to reduce levels of negative cytokines in addition to the added benefit of proangiogenic factors. The mechanisms of reducing, sequestering, or limiting production of TNF-α, and other toxic products of inflammation, require more study and should be a focus of future work in cell-based therapies.
Here we also provide additional insight toward the mechanisms of enhanced late outgrowth EPC function beyond survival, namely improved adhesion and migration. Our in vitro observations of enhanced ECM adhesion and migration toward human cardiomyocytes or other EPCs under stress account, at least in part, for the greater integration of transplanted cells in vivo after MI. The mechanisms by which Akt/HO-1 EPCs achieve enhanced adhesion through binding the ECM remain to be established. However, E-selectin levels were elevated in Akt/HO-1 EPCs (data not shown), which is known to augment EPC function (30). Promoting migration for neovessel formation through paracrine communication between late outgrowth EPCs and highly stressed human cardiomyocytes is intriguing. Likely, this is mediated by a combination of autocrine/paracrine factors, such as the chemokine (C-C motif) ligand 3 (CCL3) and CCL4, which were significantly higher in Akt/HO-1 EPCs (Table 1). The role of these molecules may have been undervalued as bystanders of inflammation since they are better known for their role in leukocyte trafficking. Infarct recovery requires overlapping steps of inflammation, repair, and perhaps even limited regeneration in the remodeling processes, much of it requiring autocrine/paracrine coordination. However, identifying the complex interplay of chemokine signaling is a challenge (22), particularly in vivo, and may require advanced in vivo imaging techniques. Future studies are needed to further identify the detailed intercellular signaling interactions between progenitors and differentiated cells, particularly after injury.
Our study focused on human late outgrowth EPCs, but our results are in agreement with other investigations implicating Akt and HO-1 in rodent MSC function and ischemic recovery. Our secretome findings are also supported by previous work, which revealed the importance of factors such as IL-8, platelet-derived growth factor (PDGF), and HGF (10,22,44,45). However, our secretome data are not in complete agreement with findings identifying the importance of SDF1-α and vascular endothelial growth factor (VEGF) (22–25). For example, at the protein level, SDF1-α and VEGF did not change dramatically in our study. Though this does not suggest that those factors are irrelevant, it could suggest that different paracrine factors are released by different cells or that differences are specific to human cells compared to rodent cells. Importantly, our study has revealed the proof-in-principle for added benefit of using Akt/HO-1 gene expression to enhance human late outgrowth EPC-based therapy after MI. Targeting key upstream intracellular mechanisms such as Akt/HO-1 confers pleiotropic benefits to the cell, its function, and its capacity to modify the extracellular environment. We have demonstrated that the benefits conferred are both intrinsic (i.e., reduced oxidative stress) and extrinsic (i.e., reduced TNF-α). Combined expression of Akt and HO-1 in late outgrowth EPCs promotes their ability to withstand multifactor stresses that cells would be exposed to in the post-MI environment. Enhanced Akt and HO-1 expression in EPCs confers heightened function through improved adhesion, migration, and conditioning of the hostile microenvironment, allowing greater neovessel formation and promoting host cell survival. Provided that we can achieve a clinically safe means of gene delivery, gene therapy of EPCs ex vivo could be an option for rejuvenating patient cells or further enhancing tissue engineering (33) to improve survival and function in the infarct microenvironment or for enhancing the clinical utility of EPCs in patients with few EPCs as a consequence of cardiovascular risk factors.
Footnotes
Acknowledgments
This work was funded in part by grants from the Canadian Institutes of Health Research (MOP79506 to L.G.M. and C.A.W.; MOP79459 and MOP93689 to C.D.F.) and the Heart and Stroke Foundation of Ontario (NA5779 to L.G.M. and C.A.W.; T6604 to R.K.L.). K.R.B. and R.A.D. were recipients of CIHR/HSF G.R.E.A.T. program doctoral awards. K.R.B. is a Heart and Stroke Foundation of Canada research fellow. D.P. was a recipient of the Deutsche Forschungs-Gemeinschaft postdoctoral fellowship. C.D.F. holds a Canada Research Chair in molecular, cellular, and physiological medicine. R.K.L. holds a Canada Research Chair in cardiac regeneration. The authors declare no conflict of interest.