
Abstract
Therapeutic effects of skeletal myoblast transplantation into the myocardium are mediated via paracrine factors. We investigated the ability of myoblast-derived soluble mediators to protect cardiomyocytes from oxidative stress. Fetal rat cardiac cells were treated with conditioned medium from cultures of myoblasts or cardiac fibroblasts, and oxidative stress was induced with H2O2. Myoblast-derived factors effectively prevented oxidative stress-induced cardiac cell death and loss of mitochondrial membrane potential. This protective effect was mediated via epidermal growth factor (EGF) receptor and c-Met signaling, and mimicked by neuregulin 1 but not EGF. Microarray analysis of cardiac cells treated with myoblast versus cardiac fibroblast-derived mediators revealed differential regulation of genes associated with antioxidative effects: cystathionine-γ-lyase (est), xanthine oxidase, and thioredoxin-interacting protein as well as nibbles homolog 3 (trib3). Cardiac cell pretreatment with tunicamycin, an inducer of trib3, also protected them against H2O2-induced cell death. Epicardial transplantation of myoblast sheets in a rat model of acute myocardial infarction was used to evaluate the expression of CST and trib3 as markers of myoblasts' paracrine effect in vivo. Myoblast sheets induced expression of the CST as well as trib3 in infarcted myocardium. CST localized around blood vessels, suggesting smooth muscle cell localization. Our results provide a deeper molecular insight into the therapeutic mechanisms of myoblast-derived paracrine signaling in cardiac cells and suggest that myoblast transplantation therapy may prevent oxidative stress-induced cardiac deterioration and progression of heart failure.
Keywords
Introduction
Cardiac ischemic insults, such as infarction or ischemia-reperfusion, pathologically alter the myocardial oxygen balance. The ensuing cell death has been associated with both endoplasmic reticulum (ER) stress and with high-level production of reactive oxygen species (ROS) that are uncompensated by cellular antioxidant protection (46). Progressive cardiac cell death leads to myocardial dysfunction and heart failure (5,12). One of the key enzymes in cellular adaptation to ER and oxidative stress is cystathionine-γ-lyase (CST) (8,17). CST is an enzyme of the reverse trans-sulfuration pathway that catalyzes H2S-producing reactions from homocysteine and cysteine (17,36,50). H2S, on the other hand, is a gaseous mediator that has antioxidative effects and mediates the cardioprotective effects of CST (4,36,50). Moreover, CST has been shown to contribute to mitochondrial protection and stabilization of energy production especially under stress conditions (10).
Transplantation of skeletal myoblasts has been suggested to inhibit cardiomyocyte death (40) and to facilitate cardiac repair after myocardial infarction (MI)-induced injury (30,39). The molecular mechanisms mediating these beneficial effects are, however, poorly understood. By proper choice of the delivery route, any adverse effects associated with myoblast transplantation, such as needle-induced tissue injury and arrhythmias (22), can be avoided (30). For example, epicardially transplanted myoblast sheets effectively mediate the beneficial paracrine effect shown to inhibit ventricular remodeling, to suppress fibrosis, and to induce therapeutic angiogenesis (18) without causing arrhythmias (22,30). Several mediators including vascular endothelial growth factor (VEGF) (18,31), placental growth factor (PLGF) (18), and hepatocyte growth factor (HGF) (26,27,31) may contribute to these paracrine therapeutic actions. Interestingly, ligandmediated activation of VEGFR (by VEGF and PLGF) or c-Met (by HGF) has been shown to protect cardiomyocytes from cell death (35). In addition, signaling via the epidermal growth factor receptor (EGFR) pathway has been shown to counteract ROS-mediated damage in cardiomyocytes (37).
Our aim was to investigate if myoblast-mediated signals can induce protection against oxidative stress in cardiac cells and to elucidate the mechanisms and marker genes associated with such protection. To this end, we pretreated cultures of primary rat fetal cardiac cells with conditioned medium from myoblasts and then induced oxidative stress with H2O2. We then evaluated cardiac cell viability and the persistence of mitochondrial membrane potential. We investigated the contributions of EGFR, VEGFR, and c-Met receptor tyrosine kinase pathways using pathway-specific small molecule inhibitors. Moreover, we performed a transcriptome level analysis of gene expression of cardiac cells pretreated with myoblast- or cardiac fibroblast-conditioned medium to discover those pathways specifically associated with myoblast-derived paracrine factor-induced antioxidative action. To verify that a similar myocardial response to myoblast transplantation exists in vivo, we evaluated the expression of marker genes, as found in microarrays, in a rat model of MI with or without myoblast sheet transplantation therapy.
Materials and Methods
Isolation of Cardiac Cells and Cell Culture
Cardiac cells were isolated from fetal Wistar rats (E17.5, Harlan Laboratories, Rossdorf, Germany). These animals were euthanized by decapitation and hearts enzymatically digested with collagenase IV (1 mg/ml, Worthington Biomedical, Lakewood, NJ, USA) and trypsin (2 mg/ml; Sigma-Aldrich, St. Louis, MO, USA). After digestion, cells were plated in DMEM containing 10% horse serum and 5% fetal bovine serum (all from Life Technologies, Paisley, UK) for 90 min to allow attachment of fibroblasts. The nonadherent cell population was collected and replated for experiments. To initiate a purified cardiac fibroblast culture, the adherent cell population was passaged four times in DMEM containing 10% fetal bovine serum prior to experiments.
The L6 skeletal myoblast cell line came from the American Type Culture Collection (ATCC-1458; Manassas, VA, USA). It was cultured in DMEM containing 10% fetal bovine serum, and the cells were passaged three times per week to prevent myoblast fusion and differentiation. To acquire myoblast-conditioned medium (MCM) and cardiac fibroblast-conditioned medium (CFCM) for experiments, the cells were grown in six-well plates (Nalge Nunc International, Roskilde, Denmark) to confluency. The monolayers were thoroughly washed to remove any serum remnants. Culture medium was replaced with 1 ml serum-free DMEM per well; 24 h later, conditioned medium was collected for experiments.
Culture Treatments MTTAssay, Cytometry, and Caspase 3 Activity Assay
To determine the ability of myoblast-secreted paracrine factors to protect cardiac cells from oxidative stress, cardiac cells were plated on 96-well plates (Nalge Nunc International) and were allowed to adhere for 48 h. Culture medium was then changed to fresh serum-free DMEM, MCM, or CFCM. For some experiments, cultures were treated with recombinant growth factors (100 ng/ml unless indicated otherwise): rat epidermal growth factor (EGF), human neuregulin 1 (NRG1), and human HGF (all from Peprotech, Rocky Hill, NJ, USA). To induce trib3 expression in cardiac cells, cells were treated with tunicamycin (1-300 ng/ml; Sigma-Aldrich), an inhibitor of N-linked glycosylation in the ER. Tunicamycin has also been associated with cardiomyocyte protection (52). Because tunicamycin is also associated with induction of ER stress and cardiomyocyte apoptosis (23), several concentrations were applied. These treatments were continued for 24 h, after which cultures were washed, and the indicated concentration of H2O2 (Sigma-Aldrich) was added to the cultures to induce oxidative stress and cell death; 24 h later, cardiac cell viability was determined with the MTT (Sigma-Aldrich) assay (45). The formazan converted by living cells was dissolved in DMSO (Sigma-Aldrich) and quantitated with the Labsystems Multiscan RC plate reader (Thermo Fisher Scientific, Vantaa, Finland) at λ540-λ670.
In some experiments, the preconditioning and H2O2 treatment steps were performed in the presence of small molecule receptor tyrosine kinase inhibitors. AG1478 (ErbB), SU11274 (c-Met), and SU5416 (VERFR1/2) all came from Sigma-Aldrich and were used at 0.2-1 μM.
To analyze the fractions of viable, injured, and dead cells after H2O2 stress, treated cells were trypsinized and dissociated into single-cell suspension, and 1 × 106 cells/ ml in phosphate-buffered saline (PBS) were stained with 10 ng/ml propidium iodide (PI) nucleic acid stain (Life Technologies) for 15 min on ice. Viable cells can exclude this dye rendering the cells unstained, whereas injured cells stain weakly and dead cells strongly, yielding three distinct populations. Samples were immediately analyzed with the Accuri C6 cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data analysis was with the BD Accuri C6 Analysis Software (BD Biosciences).
As a biochemical assay for H2O2-induced apoptosis, caspase 3 activity was measured in cultures using the EnzChek Caspase-3 Assay Kit (Z-DEV-AMC substrate, Life Technologies). Apoptosis was induced for 6 h (100 μM H2O2), and samples were processed according to the kit protocol. Caspase 3 activity was determined as the relative fluorescence of the cleaved substrate. Data were acquired with the Wallac Victor2 (Perkin-Elmer, Wellesley, MA, USA) and normalized to control samples without H2O2 treatment.
Mitochondrial Membrane Potential
Because loss of mitochondrial membrane polarity is an early marker of cardiomyocyte death (47), we determined whether myoblast-secreted mediators are able to preserve mitochondrial membrane polarity. Cardiac cell cultures were pretreated with MCM, CFCM, or fresh culture medium for 24 h before inducing oxidative stress with H2O2 (100 μM) for another 24 h. Then cultures were treated with 100 nM tetramethylrhodamine ethyl ester (TMRE; Sigma-Aldrich) for 15 min and washed thereafter to remove any excess dye. TMRE diffuses to functional mitochondria where it exhibits bright fluorescence. This signal is quenched upon opening of the mitochondrial transition pore and the subsequent loss of mitochondrial membrane potential. Fluorescence signal was quantified with Wallac Victor2, and fluorescence images were acquired with an inverted fluorescent microscope (Olympus, Tokyo, Japan)
Western Blotting
Western blotting was performed to analyze the ability of NRG1, MCM, and CFCM to induce phosphorylation of the ErbB4 receptor. Cardiac cell cultures were treated with either NRG1 (100 ng/ml), MCM, or CFCM for 10 min. Control cultures were treated with fresh medium. Then cells were collected, lysed into Laemmli Sample Buffer (Bio-Rad, Hertfordshire, UK), and separated using polyacrylamide gel electrophoresis (Bio-Rad). Proteins were then transferred into Whatman nitrocellulose membranes (Sigma-Aldrich) and blocked in 5% bovine serum albumin (Sigma-Aldrich). For protein detection, blots were incubated with anti-phospho-ErbB4 (Tyr984, 1:100 dilution; Cell Signaling Technology, Danvers, MA, USA) or anti-Pan Actin (1:500; Thermo Fisher Scientific). After washing, blots were incubated with fluorophoreconjugated IRDye secondary antibodies (goat anti-rabbit 800CW and goat anti-mouse 680LT; 1:10,000; Li-Cor, Lincoln, NE, USA). Fluorescence signal was detected with Odyssey reader (Li-Cor).
Animals
To study the impact of myoblast-derived factors on cardiac gene and protein expression in adult rats, myoblast cell sheets were engineered utilizing temperature-responsive cell culture dishes (UpCell™; CellSeed, Tokyo, Japan). These dishes are grafted with a polymer that at room temperature allows spontaneous cell detachment without any enzyme digestion or mechanical force, and 6 × 106 myoblasts were plated on such 30-mm dishes where sheets formed during a 16-h incubation and sheets were harvested for transplantation.
Male Wistar rats (8 weeks old; Harlan Laboratories) were anesthetized with 0.4 mg/kg medetomidine SC (Orion Pharma, Turku, Finland) and 60 mg/kg ketamine (Parke-Davis, Barcelona, Spain). The animals were intubated, and the hearts were exteriorized through a left thoracotomy and pericardiotomy. Myocardial infarction was induced by ligation of the left anterior descending coronary artery ligation 3 mm from its origin. For transplantation groups, two myo-blast sheets (1.2 × 107 myoblast total) were then implanted on the left ventricular anterior wall. After the surgery, 1.0 mg/kg atipamezole hydrochloride SC (Orion Pharma) was administered to antagonize anesthesia and 0.05 mg/kg buprenorphine hydrocholoride SC (Reckitt and Colman Ltd., Hull, UK) twice per day for 3 days for postoperative analgesia. For immunofluorescence staining of CST and analysis of myocardial fibrosis, 20 animals, after having undergone myocardial infarction, were randomly divided to two groups receiving either myoblast sheet transplantation (Tx) or served as controls. Of these, eight animals in the Tx group and seven animals in the control group survived the operation. For RNA isolation, a total of six rats underwent this operation. Of these, three were sham operated and received no cell transplantation. Experimental procedures were conducted according to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the ethics committee of the HUS/ Meilahti Hospital Department of Surgery.
RNA Isolation and Microarray
To acquire the profile of gene expressional changes, microarray analysis was performed on cardiac cell cultures treated for 24 h with MCM, CFCM, or fresh culture medium. In addition, total RNA was isolated from superficial myocardial tissue samples 2 days after myoblast sheet transplantation. Samples were homogenized in a Precellys24 bead homogenizer with a Cryolys cooling system (Bertin Technologies, Montigny-le-Bretonneux, France), and further homogenization and subsequent total RNA isolation was carried out with the RNeasy Mini Kit (Qiagen, Hilden, Germany). Further processing of the samples and hybridization to the Affymetrix GeneChip Rat Genome 230 2.0 chip (cultured cells; Affymetrix, Santa Clara, CA, USA) was by Biomedicum Functional Genomics Unit (http://www.helsinki.fi/fugu/) according to the manufacturer's protocols. The data were analyzed with Genespring 11 software (Agilent, Santa Clara, CA, USA) and was normalized by the RMA algorithm. All data are in compliance with the Minimum Information about a Microarray Experiment (MIAME) guidelines (http://www.mged.org/Workgroups/MIAME/miame.html), and the raw and normalized data are deposited in the National Center for Biotechnology Information Gene Expression Omnibus database (GSE35393).
Immunohistological Stainings and Histology
Animals were euthanized, and hearts were excised and cut into four equal transverse sections of which the middle part next to the apex was fixed in 4% neutral-buffered formalin (FF Chemicals, Haukipudas, Finland) for 24 h. Samples were then embedded in paraffin (Sigma-Aldrich) and cut into 5-μm sections. To quantitate myocardial fibrosis, sections were stained with Sirius red (Sigma-Aldrich), scanned, and length of the infarcted area was measured using the ImageJ software (http://rsbweb.nih.gov/ij/).
For immunofluorescence staining, sections were deparaffinized and treated in 10 mM citrate buffer (Sigma-Aldrich) at 95°C for 2 h. Sections were then blocked in 2% bovine serum albumin (Sigma-Aldrich) for 2 h, washed, and incubated with an anti-CTH (mouse monoclonal, 1:100; Abnova, Heidelberg, Germany), anti-α smooth muscle actin (SMA, 1:100, rabbit monoclonal; Abcam, Cambridge, UK), or anti-von Willebrand factor (rabbit polyclonal, 1:100; EMD Millipore, Billerica, MA, USA) overnight at 4°C. Alexa Fluor 594 (1:1,000; Life Technologies) was used as a secondary antibody. Stained sections were mounted, and nuclei were counterstained using the Vectashield Mounting Medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). Images were acquired at 20x magnification for analysis and 40x magnification for representative images using an Axio Imager M2 (Carl Zeiss, Oberkochen, Germany). Total number of CST-positive vessels and relative area of CST-positive staining was quantified using ImageJ.
Real-Time PCR
Total RNA was treated with DNAse 1 (Sigma-Aldrich), and ImProm-II Reverse Transcription System (Promega, Madison, WI, USA) was used for mRNA reverse transcription. Real-time PCR was performed with LightCycler carousel-based system (Roche Diagnostics, Basel, Switzerland), and data were normalized to rat 18S rRNA (rps18) by the following primers: activating transcripton factor 4 (atf4) forward TATGGATGGGTTGGTCAGTG, reverse CTCATCTGGCATGGTTTCC; tribbles homolog 3 (trib3) forward CTTTTGGAACGAGAGCAAGG, reverse GGG TCTTCGTGAAAAAGGTG; and rps18 forward ACA TCCAAGGAAGGCAGCAG, reverse TTTTCGTCAC TACCTCCCCG. The samples were amplified using the FastStart DNA Master SYBR Green 1 (Roche Diagnostics).
Data Analysis
All data are presented as mean ± standard error of the mean. Differences between groups were analyzed with ANOVA followed by Bonferroni posttesting. Student's t-test was used for pairwise comparisons. A value of p < 0.05 was considered significant. Statistical analysis was with Graph Pad Prism 4.0 (GraphPad Software Inc., San Diego, CA, USA).
Results
Myoblast-Derived Paracrine Factors Protect Cardiac Cells Against H2O2-Induced Oxidative Stress
To study the paracrine ability of myoblasts and cardiac fibroblasts to protect cardiac cells against oxidative stress, primary rat fetal cardiac cell cultures were treated for 24 h with MCM, CFCM, or fresh culture medium followed by induction of oxidative stress with H2O2 (100 μM) for an additional 24 h (Fig. 1A). Viability and mitochondrial function of these cultures was determined with the MTT assay.

Study protocol and cardiac cell protection. (A) Cardiac cells were isolated from fetal rat hearts and cultured for 48 h. Cells were then treated with conditioned medium from myoblasts or cardiac fibroblasts for 24 h, after which microarray and real-time PCR samples were collected. Alternatively, oxidative stress was induced in cardiomyocytes by adding H2O2. (B and C) Cardiac cell viability was assessed with the MTT assay from cells treated with myoblast-conditioned medium (MCM), cardiac fibroblast-conditioned medium (CFCM), or fresh culture medium under H2O2-induced stress (B: 100 μM; C: 200 and 300 μM). (D) Cultures were treated with H2O2 (100 μM) for 24 h, incubated with propidium iodide, and analyzed by flow cytometry to differentiate between viable, injured, and dead cells. (E) Caspase 3 activity was measured in cardiac cell cultures following a 6-h treatment with H2O2. ***p < 0.001, *p < 0.05 compared to fresh control medium; †††p < 0.001, ††p < 0.01, †p < 0.05 compared to CFCM. Values represent mean ± SEM.
Evaluation of cell viability without H2O2 treatment revealed that both CFCM (absorbance λ540-λ670, 222.3 ± 11.7) and MCM (135.0 ± 3.2, p < 0.001 vs. CFCM and unconditioned medium) induced higher mitochondrial activity than did control unconditioned medium (114 ± 1.9, p < 0.001 vs. MCM and CFCM) (Fig. 1B). When oxidative stress was induced in cultures by adding H2O2, cell viability in the cultures treated with either unconditioned medium (26.4 ± 1.3, p < 0.001 vs. MCM; p < 0.05 vs. CFCM) or CFCM (47.0 ± 9.4, p < 0.001 vs. MCM) sharply declined (61% and 58%, respectively, vs. without H2O2), whereas in the cultures treated with MCM, cell viability declined only marginally (25% vs. without H2O2) (Fig. 1B). Cardiac cell viability decreased dose dependently with increasing concentrations of 200 and 300 μM H2O2. The protective effect of myoblast-secreted factors was also evident at these higher concentrations (Fig. 1C).
To determine if myoblasts secrete H2O2-neutralizing factors in the conditioned medium, the cardiac cell cultures were thoroughly washed with fresh culture medium before H2O2 treatment. Cell viability (data not shown) remained comparable to those in Figure 1B, suggesting that the protective effect is not due to direct H2O2 neutralization by the conditioned medium but rather to changes induced by myoblast-secreted factors in cardiac cells.
Cytometry analysis of propidium iodide-stained cardiac cells with or without H2O2 treatment showed a clear reduction in the fraction of viable cells (84.8% vs. 14.2%) and increase in the fractions of injured (weak staining, 2.5% vs. 59.8%) and dead cells (strong staining, 12.7% vs. 24.5%). MCM inhibited this transition effectively as evidenced by high viable fraction (79.7%), low injured fraction (3.5%), and low dead fraction (16.8%). On the contrary, these values in the CFCM group were more similar to the H2O2-treated control group (39.4%, 38.5%, 17.5%) (Fig. 1D).
The caspase 3 activity assay showed strong induction of activity in control cells by H2O2 treatment. This induction was effectively inhibited by MCM treatment (p < 0.001 vs. control; p < 0.01 vs. CFCM). CFCM group did not differ from control group in terms of caspase 3 activity.
Myoblast-Conditioned Medium Prevents H2O2-Induced Loss of Cardiac Cell Mitochondrial Membrane Polarity
Because H2O2 induces loss of mitochondrial membrane polarity, an early indicator of cell death, the ability of myoblast-secreted paracrine factors to inhibit this principal step of cardiac cell death was studied by measuring TMRE fluorescence. At 4 h after addition of H2O2 to cultures treated with CFCM there was already a marked decrease in TMRE fluorescence (25%, p < 0.001 at all time points) that continued to decrease for up to 48 h. However, in cultures pretreated with MCM, baseline TMRE fluorescence was maintained (Fig. 2A). Fluorescence images of TMRE-labeled cardiac cells show a bright red granular fluorescence in MCM-treated cells, whereas in CFCM-and control medium-treated cells, fluorescence intensity is reduced, and its localization is diffuse (Fig. 2B).

Mitochondrial membrane potential in cardiac cell protection. (A) Evaluation of the mitochondrial membrane potential in cardiac cells treated with either myoblast-conditioned medium (MCM) or cardiac fibroblast-conditioned medium (CFCM). Fluorescence intensity was determined from cardiac cells incubated with tetramethylrhodamine ethyl ester (TMRE) at indicated time points after induction of oxidative stress with H2O2 (100 μM). (B) Fluorescence images of cardiac cell cultures treated with either MCM, CFCM, or control medium. Images were acquired from cultures stained with TMRE after 24-h induction of oxidative stress with H2O2. ***p < 0.001. Values are presented as mean ± SEM.
EGFR and c-Met Signaling Mediate the Myoblast-Induced Paracrine Protection
To identify signaling pathways associated with the myoblast-mediated cardioprotective paracrine effect, small molecule receptor tyrosine kinase inhibitors were utilized to block the target cells' intracellular signaling through EGF, c-Met, or VEGF receptors. When we induced oxidative stress with H2O2 (100 μM), the MCM-induced protection was partially suppressed with both the EGFR inhibitor AG1478 (p < 0.001) and the c-Met inhibitor SU11274 (p < 0.01), but not with the VEGFR inhibitor SU5416 even at 1 μM concentrations (Fig. 3A), suggesting that the paracrine protection by skeletal myoblasts selectively utilizes EGFR and c-Met receptor signaling in cardiac cells. To verify that the inhibitors were not toxic to cardiac cell cultures, cell viability was measured in cultures treated with fresh control medium (MTT assay, 0.92 ± 0.03), DMSO vehicle (0.89 ± 0.05), AG1478 (0.90 ± 0.04), SU11274 (0.93 ± 0.03), and SU5417 (0.90 ± 0.03). For this assay, all inhibitors were used at 1 μM concentration, which was the highest one used in the experiments. Thus, the inhibitors or vehicle were not toxic to the cells.
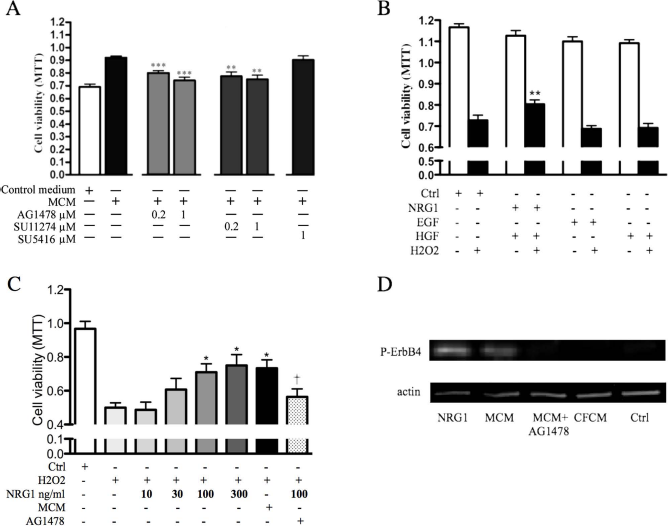
Signaling pathways of cardiac cell protection by myoblast-derived paracrine factors. (A) Cardiac cell cultures were treated with either myoblast-conditioned medium (MCM) or fresh control medium. Indicated samples were in the presence of inhibitors for epidermal growth factor (EGF) receptor (AG1478), c-Met (SU11274), and vascular endothelial growth factor receptor (SU5416). All cultures were treated with H2O2 (100 μM) to induce oxidative stress, and cell viability was measured with the MTT assay. (B) Cardiac cell cultures were treated with recombinant proteins EGF, neuregulin 1 (NRG1), or hepatocyte growth factor (HGF) for 24 h. Oxidative stress was induced as described and viability determined with MTT assay. (C) Oxidative stress was induced in cardiac cells with H2O2 with indicated concentrations of NRG1, MCM, or fresh control medium. The protective effect of NRG1 was compared to MCM. AG1478 treatment with NRG1 served as control. (D) ErbB4 receptor phosphorylation was determined with Western blotting in cardiac cell cultures by NRG1, MCM, cardiac fibroblast-conditioned medium (CFCM), and fresh control medium. The ability of AG1478 to inhibit MCM-induced phosphorylation was also determined. ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05 compared to (A) MCM treatment without inhibitors, (B and C) H2O2-treated control. †p ≤ 0.05 compared to treatment with H2O2 + 100 ng/ml NRG1 (C). Values are presented as mean ± SEM.
Next, cardiac cell cultures were treated with recombinant growth factors NRG1, EGF, and HGF (all at 100 ng/ml) to validate the EGFR- and c-Met-dependent protective response identified in the previous experiment. Only those cultures treated with NRG1 showed higher viability under oxidative stress compared to control cultures. EGF stimulation of EGFR or HGF stimulation of c-Met did not show protection against H2O2-induced oxidative stress. The result suggests that myoblast paracrine factor-mediated protection utilizes EGFR-dependent pathways related to NRG1 signaling (Fig. 3B).
To elucidate if NRG1 alone is sufficient to mimic the protective response to the same extent as MCM, a dose-response experiment was performed by treating cardiac cell cultures with different concentration of NRG1 followed by H2O2, and 100 and 300 ng/ml NRG1 was found to significantly protect cardiac cells against oxidative stress as measured by MTT assay, whereas lower concentrations were insufficient to exert protection. This protective response was inhibited by AG1478. Interestingly, the protective efficacy of NRG1 was similar to that of MCM (Fig. 3C). Western blotting showed that both NRG1 and MCM phosphorylate the ErbB4 receptor. Phosphorylation at this site (pY984) has been shown to initiate downstream signaling (38). No receptor phosphorylation was detected with cardiac cells treated with CFCM. Furthermore, MCM-induced ErbB4 phosphorylation was inhibited by treatment with AG1478 (Fig. 3D), underlining the importance of ErbB4 signaling for cardiac cell protection under H2O2 stress.
Myoblast-Conditioned Medium Induces Differential Gene Expression in Cardiac Cells
To elucidate changes in gene expression induced by myoblast-secreted factors, genome-wide gene expression profiles were compared with cardiac cells cultured in fresh culture medium, MCM, or CFCM (Table 1). Differential regulation was found of over twofold in 563 (MCM vs. culture medium) and 1,708 (MCM vs. CFCM) genes. Of these, 91 were induced or suppressed by MCM in both comparisons. Among these were upregulation of cth, a suppressor of oxidative stress and damage in the heart (4), and downregulation of two genes known to induce oxidative stress and contribute to cardiac dysfunction: thiore-doxin-interactin protein (txnip) (49) and xanthine oxidase (xdh) (14). Moreover, several upregulated genes were associated with unfolded protein response (UPR). These included trib3 (6), ddit3 (13), hspa5 (13), and atf4 (13). Furthermore, cardioprotection-associated soluble mediators such as B-type natriuretic peptide nppb (41) and brain-derived neurotropic factor (bdnf) (24) were induced. Interestingly, proinflammatory mediators such as cxcl12, which also induces oxidative stress in cardiomyocytes (2), and cc12, which contributes to cardiac reperfusion injury (32), were downregulated by MCM treatment.
Gene Expression Induced by Myoblast-Derived Paracrine Factors in Cardiac Cells
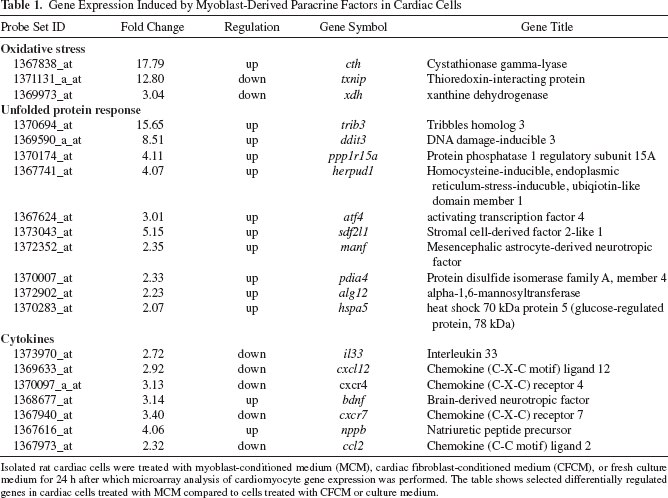
Isolated rat cardiac cells were treated with myoblast-conditioned medium (MCM), cardiac fibroblast-conditioned medium (CFCM), or fresh culture medium for 24 h after which microarray analysis of cardiomyocyte gene expression was performed. The table shows selected differentially regulated genes in cardiac cells treated with MCM compared to cells treated with CFCM or culture medium.
Tunicamycin, Inducer of trib3, Protects Cardiac Cells Against H2O2-Induced Death
Tunicamycin, an inhibitor of N-linked glycolysation, induces expression of trib3 and protects cardiomyotes against oxidative stress (52). At higher concentrations (1 μg/ml), however, tunicamycin has been shown to induce ER stress and to induce cardiomyocyte death (23). Because selective trib3 induction was evident in the microarrays as a marker of myoblast-induced cardiomyocyte protection, the ability of tunicamycin at various concentrations to modulate the cardiomyocytes' response against H2O2-induced oxidative stress was investigated. As expected, at low concentrations (9 to 30 ng/ml), tunicamycin treatment protected the cardiomyocytes against H2O2-induced stress (an 18% and 17% increase in cell viability, respectively, p < 0.05). When tunicamycin was applied at higher concentrations (100 to 300 ng/ml) the protective effect was lost, and at the highest concentration tunicamycin promoted H2O2-induced cell death (15% decrease in cell viability, p < 0.05) (Fig. 4). These results confirm the observation that tunicamycin has dose-dependent opposite effects on cardiac cell protection against oxidative stress (23,52).
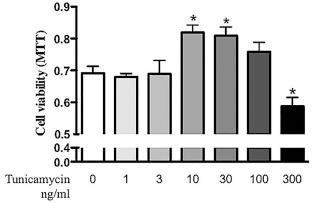
Induction of antioxidative protection by low-concentration tunicamycin and exacerbation of stress response by high-concentration tunicamycin. Cardiac cell cultures were treated with tunicamycin for 24 h to induce an unfolded protein response. Thereafter, tunicamycin was removed by washing, and oxidative stress was induced for 24 h by adding H2O2 (100 μM) in fresh medium. Thereafter, cardiac cell viability was assessed with MTT assay. *p ≤ 0.05. Values are presented as mean ± SEM.
Myoblast Sheet Transplantation Induces cst and trib3 Expression In Vivo
Next, we investigated whether the in vitro expression of cst, trib3, and atf4 associated with the protective response was also relevant in vivo. Skeletal myoblasts sheets (Tx group) were therefore epicardially transplanted after induction of MI in rats. Animals in the control group received MI without sheet transplantation. CST was detected using immunofluorescence microscopy. Increased number of CST-positive cells in the Tx group (Fig. 5A) was found. Staining of CST in infarcted hearts showed a similar staining pattern with α-SMA around blood vessels, whereas vWF staining showed signal from a single-cell layer in the vessel intima (Fig. 5B). These results thus suggest that the majority of the CST signal is derived from smooth muscle cells. Fibrotic infarct areas as well as cardiomyocytes were CST negative (data not shown). There was a 68% increase in the area of CST-positive staining in border zones of infarcted hearts in the Tx group versus control group (0.069 ± 0.010 vs. 0.041 ± 0.008%, p < 0.05) (Fig. 5C). Moreover, the Tx group had 94% higher number of positive vessels in the infarct border zone compared to the control group (5.75 ± 0.41% vs. 2.96 ± 0.29%, p < 0.001) (Fig. 5D).

Expression of cystathionase (est), tribbles homolog 3 (trib3), and activating transcription factor 4 (atf4) in vivo. Seven days after myocardial infarction with or without myoblast sheet transplantation (Tx), histological sections were prepared for immunofluorescence staining and RNA isolated from myocardium. (A) Immunoflurescence staining of CST in infarct border areas of myoblast sheet transplantation (Tx) and control groups. (B) CST staining was localized to blood vessel structures and showed a similar staining pattern to α smooth muscle actin (SMA) but not to von Willebrand factor (vWF). (C) Number of CST-positive vessels in the infarct border areas. (D) Relative area of CST-positive staining in border area. (E) Gene expression of trib3 and atf4. RNA was isolated from sham-operated hearts or myoblast sheet-implanted hearts 48 h after implantation. Expression of trib3 and atf4 in the myocardium was determined by real-time PCR and normalized to rpsl8s expression. ***p<0.001, *p<0.05. Values are presented as mean±SEM.
Analysis of gene expression revealed that trib3 as a marker of the antioxidative response was significantly upregulated by 89% in the Tx group (0.0072 ± 0.0015 vs. 0.0038 ± 0.0011, p < 0.05, values normalized to rps18S expression). Expression levels of atf4 did not show a significant difference (0.0056 ± 0.0023 vs. 0.0041 ± 0.0012) (Fig. 5D).
Therapeutic Effects of Myoblast Sheet Transplantation
Seven days posttransplantation, myoblast sheets were detectable on the infarcted hearts (Fig. 6A, black arrow). Analysis of the infarcted wall thickness showed that the Tx group had 32% thicker left ventricular wall compared to the control group (0.042 ± 0.003 vs. 0.032 ± 0.002, p < 0.01) (Fig. 6B). Moreover, analysis of myocardial fibrosis from Sirius red-stained sections showed reduced fibrosis in the Tx group (0.141 ± 0.030 vs. 0.259 ± 0.045, p < 0.05) (Fig. 6C).

Analysis of ventricular wall thickness and myocardial fibrosis. Seven days after myocardial infarction with or without myoblast sheet transplantation (Tx), histological sections were prepared and stained with hematoxylin and eosin (H&E) and Sirius red. (A) Myoblast sheet structures were evident in H&E-stained sections in the Tx group (upper panels, arrow). Sirius red staining demonstrated reduced scar area (lower panels). (B) Infarct wall thickness was significantly higher in the Tx group compared to controls. (C) Quantification of Sirius red staining indicating the amount of myocardial fibrosis showed a significant reduction in the Tx group. *p < 0.05. Values are presented as mean ± SEM.
Discussion
The therapeutic beneficial effects of skeletal myoblast transplantation are mainly attributable to the myoblast-derived paracrine factors' actions on the failing myocardium (22,28,39,44). Although these secreted mediators are recognizable as the effectors of myoblast therapy, details to their mechanisms of action still remain elusive. Because oxidative stress and overtly high production of ROS play major roles in the development of heart failure after an ischemic insult (3), our aim in this study was to clarify whether the myoblast-secreted paracrine factors could institute cardiac cell protection against oxidative stress. Intriguingly, the factors secreted from myoblasts were superior to those from cardiac fibroblasts in their ability to protect cardiac cells against oxidative stress. Previously it has been reported that paracrine factors secreted by cardiac fibroblasts can inhibit cardiomyocyte death and exert cardioprotection (29). In our data this effect was evidenced as increased viability of unstressed cardio-myocytes in response to CFCM. Although in this study CFCM gave the cardiac cells minute protection against oxidative stress, it was by far exceeded by the protective effect of paracrine mediators from skeletal myoblasts. This further strengthens the rationale for using skeletal myoblasts as the therapeutic cells of choice.
We found that inhibition of EGFR or c-Met signaling by small molecule inhibitors partially attenuated the protective effects of MCM. Moreover, pretreatment of cardiac cells by stimulation of EFGR with a recombinant ligand, NRG1, was able to award protection against H2O2-induced stress in a dose-dependent manner. Recent studies suggest an important role for the EGFR pathway in cardioprotection against oxidative stress. Inhibiting the ischemic preconditioning-associated EGFR phosphorylation attenuated the protective effect of pretreatment, suggesting that the EGFR pathway is crucial for cardiac protection against oxidative stress (48). The EGFR family can be activated by several ligands. Of these, NRG1 (16) and heparin-binding EGF (HB-EGF) (15) are expressed by myoblasts and have antioxidative function in the heart (43,48). We found that both NRG1 and MCM induce phosphorylation of the ErbB4 receptor, suggesting that this pathway is crucial for the protective response induced by skeletal myoblasts. Cardiac endothelial cells have been previously shown to exert a paracrine protective effect on cardiomyocytes (21). Interestingly, also this heart's endogenous protective effect utilized NRG1 and ErbB4 signaling. Our results thus show that skeletal myoblasts can activate a paracrine cardioprotective pathway that is intrinsically utilized by cardiac cells to guard against oxidative stress-induced myocyte loss.
Myoblasts also express matrix metalloproteinases (34, 39) that are known to cleave and thereby activate not only NRG1 but HB-EGF as well (21,42). In addition to NRG1, the EGFR pathway may therefore be activated by a matrix metalloproteinase-dependent cleavage of endogenous cardiac HB-EGF. Activation of EGFR and its potentiation by c-Met has been reported (9). Given the effective inhibition observed by the c-Met inhibitor but the inability of the recombinant HGF to give antioxidative protection, our data suggest that myoblast paracrine factor-induced protection also involves synergistic cooperation between these receptors or their intracellular signaling cascades.
Although myoblasts express VEGF and other VEGF family ligands (18,28,31) that are linked to direct protection against oxidative stress (20,51), we found no modulation of the protective effect with VEGFR1/2 inhibitor even at high 1 μM doses. This suggests that the VEGFR signaling pathway has little significance in mediating the myoblasts' protective effect in cardiac cells. Despite the fact that VEGFR activation has been shown to be crucial for the endothelial cell responses in myoblast sheet therapy (40) and that VEGF expression has been suggested to mediate the beneficial effects of bone marrow mesenchymal stem cell therapy (25), our results indicate that this pathway does not contribute to the myoblasts' direct antioxidative protection of cardiomyocytes.
Our analysis of cardiac cell gene expression in response to myoblast-secreted factors revealed potent upregulation of cst, which in the heart catalyzes L-cysteine to produce the gaseous mediator H2S that then helps to alleviate oxidative stress and protect the heart against ischemic damage (4,50). Interestingly, H2S also alleviates ER stress in the heart (7). Following transplantation to infarcted hearts, myoblast sheets induced higher expression of CST in blood vessels of the infarct border areas. Previous studies have suggested an important cardioprotective role for H2S through regulation of vasodilatation (17,19). Our results provide evidence that CST can be induced by myoblast transplantation and that CST may also play a role in cardioprotection by reducing cardiac cell death. Additionally, H2S production has been shown to reduce cardiac fibrosis following MI (33). This is in line with our results showing reduced fibrosis following myoblast transplantation therapy. Our results demonstrate a novel therapeutic mechanism of myoblast transplantation by induction of higher CST expression in the heart. In addition to CST, we found downregulation of txnip, an inhibitor of the antioxidative and cardioprotective factor thioredoxin (49). Furthermore, the pro-oxidative gene xdh that contributes to cardiac dysfunction (14) was also downregulated. These gene expression changes related to promotion of cellular antioxidant defenses can further help explain the protective effects of skeletal myoblasts.
Oxidative stress-induced ER stress has been associated with promotion of cardiomyocyte death. UPR is induced as an adaptive response to ER stress and is suggested to be cardioprotective (42). As the most upregulated gene—specifically responding to myoblast therapy—we indentified trib3, a pseudokinase and a modulator of intracellular signaling associated with the UPR (6). Interestingly, our microarray data reveal a number of genes associated with the UPR in cardiomyocytes. Our experiment utilizing tunicamycin as an inducer of UPR and trib3 demonstrates that at low tunicamycin concentrations, UPR/trib3 induction is protective against oxidative stress but this effect is lost at higher concentrations. Our data support the finding of an association of tunicamycin-induced UPR with cardiomyocyte protection (52). In contrast to our data, trib3 expression in the heart has been associated earlier with promotion of apoptosis (1). In this study, trib3, as a part UPR, is associated with cardiomyocyte protection against oxidative stress. Interestingly, a recent report described downstream activation from EGFR tyrosine kinases of ER stress in diabetes (11). Our results indicate existence of a similar link between EGFR and ER stress utilizing trib3 expression as a marker or effector gene. We show that the ER stress response activated by myoblast-derived paracrine factors, at least partially involving EGFR signaling, serves a protective function against oxidative stress in cardiac cells. Furthermore, our results suggest that cst may mediate this effect.
In this study, we provide evidence that skeletal myoblast-secreted factors can induce an oxidant damageresistant phenotype on cardiac cells. This phenotype, associated with expression of cst and trib3, was inducible also in vivo. Intriguingly, the myoblasts' paracrine actions were superior to those of cardiac fibroblasts that represent signals from the physiological cardiac mesenchyme. Our results thus suggest that skeletal myoblast transplantation to a failing ischemic heart can help the remaining functional myocytes to survive and may thus prevent cell death associated with disease progression.
Footnotes
Acknowledgments
This work was supported by Core-to-Core funding between the Academy of Finland and the Japanese Society for Promotion of Science (JSPS) (grant No. 120313); RAM project (grant No. 40102/08) between Japan Science and Technology Agency (JST) and the Finnish Funding Agency for Technology and Innovation (TEKES); and the Finnish Foundation for Cardiovascular Research. We thank Lahja Eurajoki for the expertise with in vitro studies; Mayuu Kataoka for the help with histological stainings; and Olli Valtonen, Veikko Huusko, and Virpi Norppo for animal care. Professor Dan Lindholm kindly provided the rats for cardiac cell isolation. This work was carried out at Pharmacology, University of Helsinki, and at the Department of Cardiovascular Surgery, Osaka Graduate School of Medicine. The authors declare no conflicts of interest.