
Abstract
Irisflorentin is an isoflavone component derived from the roots of Belamcanda chinensis (L.) DC. In traditional Chinese medicine, this herb has pharmacological properties to treat inflammatory disorders. Dendritic cells (DCs) are crucial modulators for the development of optimal T-cell immunity and maintenance of tolerance. Aberrant activation of DCs can induce harmful immune responses, and so agents that effectively improve DC properties have great clinical value. We herein investigated the effects of irisflorentin on lipopolysaccharide (LPS)-stimulated maturation of mouse bone marrow-derived DCs in vitro and in the contact hypersensitivity response (CHSR) in vivo. Our results demonstrated that treatment with up to 40 μM irisflorentin does not cause cellular toxicity. Irisflorentin significantly lessened the proinflammatory cytokine production (tumor necrosis factor-α, interleukin-6, and interleukin-12p70) by LPS-stimulated DCs. Irisflorentin also inhibited the expression of LPS-induced major histocompatibility complex class II and costimulatory molecules (CD40 and CD86) on LPS-stimulated DCs. In addition, irisflorentin diminished LPS-stimulated DC-elicited allogeneic T-cell proliferation. Furthermore, irisflorentin significantly interfered with LPS-induced activation of IκB kinase, c-Jun N-terminal kinase, and p38, as well as the nuclear translocation of NF-κB p65. Subsequently, treatment with irisflorentin obviously weakened 2,4-dinitro-1-fluorobenzene-induced delayed-type hypersensitivity. These findings suggest new insights into the role of irisflorentin as an immunotherapeutic adjuvant through its capability to modulate the properties of DCs.
Keywords
Introduction
Belamcanda chinensis (L.) DC (synonym: Iris domestica), a flowering perennial herb scattered over the region of East Asia, is valuable in Chinese villages for its medicinal uses. The dried rhizome of this herb, Belamcandae Rhizoma (Shegan in Chinese), has been reported for treatment of cough, pharyngitis, asthma, swollen spleen and liver, malaria, gonorrhea, and arrow poisoning (36). Isoflavones are the major bioactive constituents of the rhizome of B. chinensis (23,38), demonstrating physiological activities that have antiinflammatory, antioxidant, anticancer, and hypoglycemic properties (19,35). Irisflorentin (Fig. 1) is the abundant isoflavone component derived from B. chinensis herbs (39). It has been shown to inhibit the inflammatory activity of lipopolysaccharide (LPS)-stimulated RAW 264.7 macrophages (12).

Chemical and molecular structure of irisflorentin.
Dendritic cells (DCs) are the most potent antigen-sensing and -presenting cells, acting to initiate and fine tune complex innate and adaptive immune responses in the body (26). DCs have been used to treat malignant tumors (2) and infection of microbial pathogens (29). In addition, DCs are considered to play a central role in inducing tolerance, predominantly through the generation of CD4+ CD25+ regulatory T-cells (7). In the absence of activation, antigen presentation by steady-state DCs might promote T-cell unresponsiveness (27). DCs have two functional phases. Immature DCs are differentiated from bone marrow progenitors and reside in the peripheral nonlymphoid tissues. These cells show high endocytic capability and low T-cell stimulation potential. Following antigen encountering, DCs process selected pathogen-derived peptides, deliver them to the surface, and change into mature DCs. They then have a reduced capacity for endocytosis/phagocytosis, and gain of competence to migrate to secondary lymphoid organs by C-C chemokine receptor type 7 expression, where they produce proinflammatory cytokines and activate naive T-cells through signaling of costimulatory molecules and the peptide/major histocompatibility complex (MHC) (24,26).
LPS of Gram-negative bacteria has been shown to promote the maturation of immature DCs and induce a protective adaptive immune response via pattern recognition receptors, such as Toll-like receptor 4 (TLR-4) (24). TLR-4 specifically associates with LPS/LPS-binding protein and CD14 coreceptor, and triggers two signaling pathways. MyD88/Toll-IL-1 resistance (TIR) domain-containing adapter protein (TIRAP) pathway (MyD88-dependent pathway) is initiated from the plasma membrane to active nuclear factor (NF)-κB and cytokine production. TIR domain-containing adapter-inducing interferon (IFN)-β (TRIF)/TRIF-related adapter molecule pathway (MyD88-independent pathway) is triggered when TLR-4 is internalized into the endosome. They activate IFN regulatory factor-3 to produce type I IFNs and CCL5 (5). A recent study found that the integrin αM (CD11b) can positively regulate both TLR-4-induced pathways on DCs and modulate the balance between innate and adaptive immunity initiated by LPS (24). Moreover, the MyD88-dependent pathway was shown to be responsible for proinflammatory cytokine expression and activation of the IκB kinase (IKK)/NF-κB pathway; three mitogen-activated protein kinase (MAPK) pathways [c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38]; and the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, all of which direct the expression of various genes linked to DC maturation (20,25).
Allergic contact dermatitis (ACD) is a prevalent inflammatory disease of the skin resulting in pruritic, eczematous lesions. It is caused by T-cell-mediated type IV delayed-type hypersensitivity responses triggered by the contact of the skin with the offending chemicals in persons who have been previously sensitized to the same chemicals (14). DCs play an important role in the initiation of ACD (17). Because DCs are central immunomodulators, controlling their activity may be a useful approach for treating ACD (3,13,16).
In the present research, we tested whether irisflorentin could affect the phenotypic and functional maturation of LPS-stimulated mouse bone marrow-derived DCs (mBM-DCs). We investigated the corresponding signaling pathways and examined the in vivo effects of irisflorentin on skin contact hypersensitivity.
Materials and Methods
Chemicals, Regents, and Antibodies
Synthesized irisflorentin (mol. wt. 386.35, 98% purity) was purchased from Fusol Material Co., Ltd. (Tainan, Taiwan), dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) to 100 mM, and stored at −20°C as a master stock solution. Roswell Park Memorial Institute (RPMI)-1640 medium, l-glutamine, fetal bovine serum (FBS), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethane-sulfonic acid (HEPES; pH 7.4), sodium pyruvate, penicillin–streptomycin, nonessential amino acid, and β-mercaptoethanol were purchased from Invitrogen (Grand Island, NY, USA). Recombinant mouse granulocyte-macrophage colony-stimulating factor (rmGM-CSF) and recombinant mouse interleukin-4 (rmIL-4) were purchased from Prospec (Ness-Ziona, Israel). LPS (from Escherichia coli 055:B5), 2.4-dinitro-1-fluorobenzene (DNFB), and other chemicals were purchased from Sigma-Aldrich unless specified otherwise. Unlabeled anti-CD16/CD32 antibody, phycoerythrin (PE)-conjugated antibody to mouse CD11c, FITC-conjugated antibody to mouse MHC class II, CD40, CD86, and isotype-matched control antibodies were purchased from GenWay Biotech (San Diego, CA, USA). The antibodies for IKKα/β, IκBα, JNK, ERK1/2, p38, Akt, their phosphorylated forms, and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). HRP-labeled secondary antibodies were purchased from PerkinElmer Life Sciences (Boston, MA, USA).
mBM-DC Generation
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Institutional Animals of China Medical University and the Care and Use of Laboratory Animals of the National Institutes of Health. All animal experiments were conducted according to a protocol approved by the Institutional Animal Care and Use Committee of the China Medical University (Permit Number: 101–32-N). Four male C57BL/6 mice (8 weeks old, 20 g, from the National Laboratory Animal Center, Taiwan) were maintained in a specific pathogen-free area at the Animal Center of China Medical University (Taichung, Taiwan) and housed in temperature-controlled rooms with a 12-h light/dark cycle and were given food and water ad libitum. mBM-DCs were acquired as described previously (31), with minor modifications (10,11). Briefly, femoral and tibial bone marrow cells of mice were first flushed and passed through a nylon mesh. Red blood cells were lysed with ACK lysis buffer (pH 7.2–7.4, 0.15 M NH4Cl, 1.0 M KHCO3, 0.1 mM Na2EDTA). Then 1 × 106 cells were placed in 24-well plates (Corning Inc., Corning, NY, USA) in 1 ml RPMI-1640 medium supplemented with 2 mM l-glutamine, 10% (v/v) heat-inactivated FBS, nonessential amino acid, 1 mM sodium pyruvate, 10 mM HEPES, rmGM-CSF (20 ng/ml), rmIL-4 (20 ng/ml), 50 μM β-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C, 5% CO2. On day 3, 1 ml complete RPMI-1640 medium containing rmGM-CSF and rmIL-4 was added to the plates to give a total of 2 ml. On days 5 and 7, 1 ml of the cell-free supernatant was exchanged, and 1 ml fresh medium containing rmGM-CSF and rmIL-4 was added. On day 8 of culture, nonadherent and loosely adherent cells (immature mBM-DCs) were collected and used for all experiments. The percentage of CD11c+ cells averaged 80% when detected by BD LSR II flow cytometry (BD Biosciences, San Jose, CA, USA).
Cytotoxicity Assay
mBM-DCs were pretreated with irisflorentin at different concentrations for 24 h and then stimulated with or without 100 ng/ml LPS for 20 h. The final concentration of DMSO in all irisflorentin-treated cultures was 0.1% (v/v) and had no effect on mBM-DCs in all experiments (data not shown). Cells were then harvested and stained by using 5 μg/ml propidium iodide (Sigma-Aldrich) or Annexin V kit (Invitrogen) according to the manufacturer's protocol. Cell viability and apoptosis were analyzed by BD LSR II flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Cell Treatment and Stimulation
Based on data of the cytotoxicity assay, mBM-DCs were pretreated with 20 or 40 μM irisflorentin for 1 h. DMSO in all irisflorentin-treated cultures with noncytotoxic concentration was 0.1% (v/v). After 1 h of incubation, the cells were washed twice using phosphate-buffered saline (PBS; Amresco, Solon, OH, USA), followed by stimulation with 100 ng/ml LPS for the indicated time points. Media and cells were collected for subsequent evaluation of DC activation and animal model. Three replicates were included in each experiment.
Cytokine Secretion Assay
Irisflorentin-pretreated mBM-DCs were cultured in six-well plates (Corning) and stimulated with LPS. After 20 h (12 h for TNF-α), the supernatants were collected, centrifuged at 13,000 rpm, and stored at −80°C. The secretions of TNF-α, IL-6, and IL-12p70 were then measured by enzyme-linked immunosorbent assay (ELISA) kits purchased from Invitrogen. The cytokine concentration was evaluated according to the manufacturer's protocol.
Flow Cytometric Analysis
Irisflorentin-pretreated mBM-DCs were stimulated with LPS for 20 h. The expression of surface molecules on DCs was determined by flow cytometry as described previously (9). Cells were harvested and resuspended in ice-cold FACS washing buffer (0.1% sodium azide and 2% FBS in PBS). Cells were then blocked with unlabeled anti-CD16/CD32 antibody (10 μg/ml, final concentration) for 10 min at room temperature and stained with FITC-conjugated anti-MHC class II (5 μg/ml, final concentration), anti-CD40 (5 μg/ml, final concentration), and anti-CD86 (5 μg/ml, final concentration) with PE-conjugated anti-CD11c (5 μg/ml, final concentration) antibody for 30 min at 4°C in the dark. The appropriated isotype-matched antibodies were used as negative controls. Stained cells were washed and analyzed by flow cytometry using FlowJo version 7.6 software (TreeStar, Inc., Ashland, OR, USA). The data were collected for 1 × 104 cells per sample. Results are expressed as the percentage of positive cells or as mean fluorescence intensity.
Allogeneic Mixed Lymphocyte Reaction Assay
Splenocytes from the spleens of sacrificed C57BL/6 mice (8 weeks old) were harvested under sterile conditions and passaged through 70-mm cell strainers. Red cells were depleted by chemical lysis using ACK buffer. Responder T-cells were isolated by using a mouse T-cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Over 90% of cells expressed CD3+, as determined by the FITC-conjugated CD3 antibody (5 μg/ml, final concentration; GenWay Biotech) using BDLSR II flow cytometry (BD Biosciences). The cell suspension was then washed twice with PBS for 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) labeling. CFSE labeling of responder cells was undertaken as previously described (6,37). The cells were resuspended in 1 μM CFSE (Molecular Probes, Eugene, OR, USA) in PBS and periodically shaken at room temperature for 10 min. The labeling process was quenched by washing once in pure heat-inactivated FBS and twice in PBS with 10% FBS. Irisflorentin-pretreated mBM-DCs were stimulated with LPS for 20 h. Cells were then harvested, washed, and were then resuspended in culture medium. Finally, the cells were mixed with the prepared responder T-cells in a ratio of 1:10 and 1:50 in U-bottomed 96-well culture plates (Corning Inc.) at 37°C, 5% CO2. A negative control (responder T-cells alone) was created for each experiment. After 3 days, the cells were harvested, washed in PBS, and T-cell proliferation was measured by flow cytometric analysis of CFSE dilution.
NF-κB Transcription Factor Assay
Irisflorentin-pretreated mBM-DCs were stimulated with LPS for 40 min. The nuclear protein fraction of cells was obtained by using the Nuclear Extraction Kit (Affymetrix-Panomics, Santa Clara, CA, USA). Sample protein concentrations were measured using RC DC Protein Assay Kit (Bio-Rad Life Science, Hercules, CA, USA). NF-κB p65 binding activity was detected with the Universal EZ-TFA Transcription Factor Assay Colorimetric kit according to the manufacturer's protocol (Millipore, Billerica, MA, USA). Briefly, nuclear extracts were incubated with biotinylated capture probes that contained the consensus sequence for NF-κB p65. To determine the specificity of NF-κB p65 DNA binding, a competitor probe, that contained the same sequence as the capture probe, but did not include any biotin modifications, was mixed with the capture probe, and also incubated in streptavidin-coated plates (Millipore). The NF-κB p65-bound biotinylated oligonucleotide was then immobilized on the streptavidin-coated plate, and the unbound material was washed away. The bound transcription factor was then determined by incubation with a rabbit anti-NF-κB p65 antibody (1:500) and a horseradish peroxidase (HRP)-conjugated secondary antibody (1:500). A negative control containing free probe and binding buffer without cell lysate was added in each assay. HRP activity was colorimetrically quantified at 450 nm using a SpectraMax M2 Microplate reader (Molecular Devices, Silicon Valley, CA, USA).
Immunofluorescence Analysis
Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 15 min at room temperature, then permeabilized with 0.3% Triton X-100 (Sigma-Aldrich) for 20 min. After washes with PBS, cells were blocked with 10% normal goat serum (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 30 min. NF-κB p65 subcellular location was determined by immunostaining with rabbit anti-mouse NF-κB p65 antibodies (1:200 dilution; Santa Cruz Biotechnology) overnight at 4°C, then with a secondary antibody, Alexa Fluor-488-conjugated goat anti-rabbit IgG (1:100 dilution; Santa Cruz Biotechnology), for 1 h at room temperature. Nuclei were counterstained with the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI, 2.5 μg/ml, final concentration; Sigma-Aldrich) for 5 min. Fluorescence signals were observed under Axio Observer inverted fluorescence microscope (Carl Zeiss MicroImaging GmbH, Göttingen, Germany).
Immunoblot Analysis
Irisflorentin-pretreated mBM-DCs were stimulated with LPS for 40 min. For use in cell lysis, modified RIPA buffer (Millipore) containing 1 mM PMSF, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 μg/ml aprotinin, 1μg/ml leupeptin, and 1 μg/ml pepstatin was used to extract proteins. The protein level of whole cell lysates was directly quantitated by using the RC DC Protein Assay Kit (Bio-Rad Life Science). Fifty micrograms of protein per sample was denatured in 6× sample buffer, loaded onto 10–12.5% sodium dodecyl sulfate (Sigma-Aldrich)-polyacrylamide (Amresco) gel electrophoresis (SDS-PAGE). After electrophoresis, proteins were transferred onto PVDF membranes (Millipore) followed by blocking with 5% nonfat milk (w/v) (Bio-Rad Life Science) for 1 h at room temperature. The membranes were then incubated with specific primary antibodies for IKKα/β (4 μg/ml, final concentration), IκBα (4 μg/ml, final concentration), JNK (2 μg/ml, final concentration), ERK1/2 (2 μg/ml, final concentration), p38 (2 μg/ml, final concentration), Akt (2 μg/ml, final concentration), and their phosphorylated forms (4 μg/ml, final concentration) overnight at 4°C (9,10). After they were washed, the membranes were incubated with appropriate secondary HRP-conjugated antibodies (1 μg/ml, final concentration). The blots were developed using Amersham enhanced chemiluminescence system (Piscataway, NJ, USA). Signals were detected by using a UVP BioSpectrum Imaging System (Upland, CA, USA).
Inhibitor Assay
IKK-2 inhibitor IKK-16, JNK inhibitor SP600125, and p38 MAPK inhibitor SB202190 were purchased from SelleckChem (Houston, TX, USA) and were dissolved in DMSO. Inhibitors were titrated at concentrations ranging from 1 to 50 μM. A dose of 30 μM for IKK-16, 60 μM for SP600125, or 60 μM for SB202190 was used for experiments, as this was the highest concentration that was not cytotoxic. For inhibitor assay, mBM-DCs were preincubated with the indicated inhibitors for 30 min and then incubated with or without 40 μM irisflorentin. After 1-h incubation, the mBM-DCs were stimulated with 100 ng/ml LPS for 20 h. Inhibitors were removed by twice washing cells with 2% FBS/PBS. mBM-DCs were collected, and an allogenic mixed lymphocyte reaction assay was performed.
Contact Hypersensitivity Test
Contact hypersensitivity (CHS) test induced by DNFB (Sigma-Aldrich) was described previously (10). In the modified protocol, the shaved abdomens of mice in each group were painted with 20 μl of vehicle (acetone/olive oil = 4:1), 0.5% (w/v) DNFB, 0.5% DNFB plus 0.1% (v/v) DMSO, or 0.5% DNFB plus irisflorentin (100 μg) for sensitization. After 6 days, the baseline shaved ear thickness of mice in each group was measured. Then mice in each group were challenged by painting on the backs of ears with 10 μl 0.2% DNFB (~1-cm diameter). Mice that were not sensitized, but were challenged with DNFB, served as negative controls. Ear thickness was measured 24 h later. Swelling of the ear was calculated by subtracting the thickness before challenge. CHS response also was evaluated by hematoxylin and eosin (Sigma-Aldrich) staining.
Statistics
All statistical analyses are expressed as mean ± standard deviation (SD) from three independent tests unless otherwise noted. Three replicates were done for each test. The differences between two means were determined by Bonferroni-corrected Student's t-test. Values of p < 0.05 were determined to be statistically significant.
Results
The Cytotoxicity of Irisflorentin
In the current study, the immunomodulatory effects of irisflorentin were tested by using mBM-DCs. Flow cytometry assays using propidium iodide and annexin V-fluorescein staining were employed to evaluate the cytotoxicity of irisflorentin. After treatments with irisflorentin at 5, 10, 20, and 40 μM for 24 h, LPS-stimulated and unstimulated DCs did not show any necrosis or apoptosis by propidium iodide staining and annexin V assays (Fig. 2). In the experiments following this test, cells were treated with irisflorentin at concentrations of up to 40 μM.

The cytotoxicity of irisflorentin on mBM-DCs. The mBM-DCs were pretreated with serially diluted irisflorentin for 24 h and then stimulated with or without LPS (100 ng/ml) for 20 h. The viability of cells was determined by propidium iodide staining and flow cytometry analysis (A). The apoptosis of cells was determined by annexin V-fluorescein staining and flow cytometry analysis (B), as described in the text. The data represent the mean ± SD (n = 3). An asterisk (*) shows significant differences between irisflorentin-untreated control samples and irisflorentin-treated samples (**p < 0.01). DMSO, dimethyl sulfoxide (vehicle).
Irisflorentin Inhibited LPS-Induced TNF-α, IL-6, and IL-12p70 Secretion
During activation and maturation, DCs shift their phenotype and functional properties. TNF-α, IL-6, and IL-12 are three important proinflammatory cytokines that induce the expression of costimulatory/accessory molecules on DCs and augment DC-mediated T-cell responses (2). Cytokine secretion was analyzed by ELISA. In unstimulated mBM-DCs, 40 μM irisflorentin did not change TNF-α, IL-6, and IL-12p70 production. Treatment with LPS caused a 27-fold (p < 0.001), 26-fold (p < 0.001), and 24-fold increase (p < 0.001) in the release of TNF-α, IL-6, and IL-12p70, respectively. Irisflorentin reduced the secretion of TNF-α, IL-6, and IL-12p70 in a concentration-dependent manner. At 40 μM irisflorentin, LPS-stimulated TNF-α secretion lessened by about 62% (p < 0.01), IL-6 release by about 52% (p < 0.01), and IL-12p70 release by about 49% (p < 0.01) (Fig. 3).

Irisflorentin inhibited TNF-α, IL-6, and IL-12p70 release in LPS-stimulated mBM-DCs. mBM-DCs were pretreated with 20 or 40 μM irisflorentin. After 1 h of incubation, the cells were washed, followed by stimulation with LPS (100 ng/ml) for 20 h (12 h for TNF-α). Media of cell culture were collected and measured for TNF-α, IL-6, and IL-12p70 levels by using an ELISA kit. The data represent the mean ± SD (n = 3). A hash (#) shows significant differences between LPS-stimulated and unstimulated cells (#p < 0.001); an asterisk (*) shows significant differences between the LPS-stimulated control samples and irisflorentin-pretreated, LPS-stimulated samples (*p < 0.05, **p < 0.01).
Irisflorentin Impaired LPS-Induced Surface Marker Expression
Connections between surface molecules on DCs and their ligands or receptors are critical for the full activation of T-cells. MHC class II associates with the antigenic peptide and then presents to CD4+ T-cells. CD40 binds CD154 on the CD4+ T-cell to acquire an activation signal, enhancing antigen presentation and the expression of other costimulatory molecules. CD86 associates with CD28 on the helper T-cell for T-cell priming and survival (26). The effect of irisflorentin on surface-specific molecules of activated DCs was measured by flow cytometry. The expression level of MHC class II, CD40, and CD86 was estimated by fluorescence intensity. Unstimulated mBM-DCs treated with irisflorentin did not alter MHC class II, CD40, and CD86 expression. LPS-stimulated mBM-DCs had significantly higher expression of MHC class II, CD40, and CD86 (p < 0.001). Treatment of 40 μM irisflorentin inhibited MHC class II (p < 0.01), CD40 (p < 0.01), and CD86 (p < 0.01) expression on mBM-DCs stimulated with LPS (Fig. 4).

Irisflorentin impaired MHC class II, CD40, and CD86 expression in LPS-stimulated mBM-DCs. mBM-DCs were pretreated with 20 or 40 μM irisflorentin. After 1 h of incubation, the cells were washed, followed by stimulation with LPS (100 ng/ml) for 20 h. The expression of MHC class II, CD40, and CD86 on CD11c+ cells was determined by flow cytometry. The data are represented as the mean fluorescent intensity ± SD (n = 3). A hash (#) shows significant differences between LPS-stimulated and unstimulated cells (#p < 0.001); an asterisk (*) shows significant differences between the LPS-stimulated control samples and irisflorentin-pretreated, LPS-stimulated samples (*p < 0.05, **p < 0.01).
Irisflorentin Abrogated Allostimulatory Capacity
To assess the effect of irisflorentin on the allostimulatory capacity of mBM-DCs, we employed a mixed lymphocyte reaction and flow cytometry assay and CFSE-labeled splenocytes from BALB/c mice as responder T-cells. As shown in Figure 5, LPS-stimulated mBM-DCs induced proliferative responses more effectively than those of the control group (p < 0.001), while irisflorentin-pretreated, LPS-stimulated mBM-DCs had low stimulatory activities compared with untreated mBM-DCs (40 μM, DCs/T-cells: 1:50, p < 0.001), demonstrating that irisflorentin treatment blocked the allostimulatory capacity of stimulated mBM-DCs.

Irisflorentin abrogated the proliferation of naive allogeneic T-lymphocytes by LPS-stimulated mBM-DCs. mBM-DCs were pretreated with 20 or 40 μM irisflorentin. After 1 h of incubation, the cells were washed, followed by stimulation with LPS (100 ng/ml) for 20 h. Finally, the cells were washed and diluted with the prepared CFSE-labeled splenocytes in a ratio of 1:10 and 1:50 in culture plates for 3 days. The proliferation of T-cells was assessed by flow cytometry. The values of LPS-stimulated DCs served as control values in the calculation of percentage of proliferation. The data represent the mean ± SD (n = 3). A hash (#) shows significant differences between LPS-stimulated and unstimulated cells (#p < 0.001); an asterisk (*) shows significant differences between the LPS-stimulated control samples and irisflorentin-pretreated, LPS-stimulated samples (*p < 0.05, **p < 0.01, ***p < 0.001).
Irisflorentin Attenuated LPS-Induced NF-κB p65 Translocation
NF-κB is a key regulator of innate immunity, and translocation of NF-κB from the cytosol to the nucleus is critical for LPS-stimulated maturation of DCs (33). Given that irisflorentin retarded the LPS activation of DCs in our research, we assayed the effects of irisflorentin on NF-κB p65 levels in the nucleus. As shown in Figure 6, LPS-stimulated DCs raised the NF-κB p65 levels in the nucleus (p < 0.001), whereas 40 μM irisflorentin treatment lowered nuclear NF-κB p65 levels in the LPS-stimulated DCs in a concentration-dependent manner (p < 0.01).
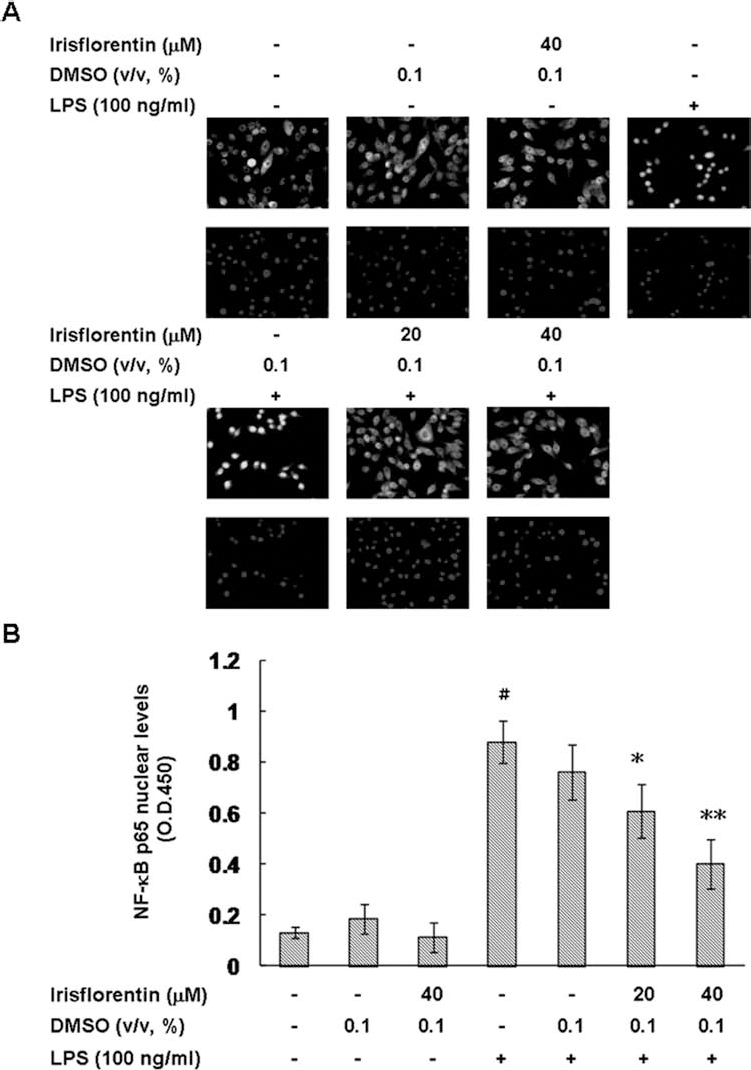
Irisflorentin attenuated the translocation of NF-κB p65 in LPS-stimulated mBM-DCs. mBM-DCs were pretreated with 20 or 40 μM irisflorentin. After 1 h of incubation, the cells were washed, followed by stimulation with LPS (100 ng/ml) for 40 min. (A) The localization of NF-κB p65 was determined by immunostaining with anti-NF-κB p65 antibody (upper image). DAPI staining shows the location of cell nucleus (lower image). Scale bar: 50 μm. (B) Cells were lysed and the nuclear fraction measured for relative binding activity of NF-κB p65 by using the Universal EZ-TFA Transcription Factor Assay Colorimetric kit. The data represent the mean ± SD (n = 3). A hash (#) shows significant differences between LPS-stimulated and unstimulated cells (#p < 0.001); an asterisk (*) shows significant differences between the LPS-stimulated control samples and irisflorentin-pretreated, LPS-stimulated samples (*p < 0.05, **p < 0.01).
Irisflorentin Decreased LPS-Induced IKK/IκB and JNK/p38 MAPK Phosphorylation in the Cytoplasm
Several signaling pathways are concerned in DC maturation (24). To further test the effect of irisflorentin in IKK/NF-κB, PI3K/Akt, and MAPK pathways, we analyzed the phosphorylation levels of major signaling factors associated with the activation of DC by using immunoblot analysis. As shown in Figure 7, LPS markedly induced IKKα/β, IκBα, JNK, and p38 phosphorylation and promoted IκBα degradation in mBM-DCs, both of which were arrested in irisflorentin-pretreated cells in a concentration-dependent manner (40 μM, p < 0.01). LPS-induced ERK1/2 and Akt phosphorylation was not blocked by irisflorentin treatment. Moreover, retarding the IKK, JNK, and p38 MAPK activities with IKK-16, SP600125, and SB202190 inhibitors, respectively, completely abolished the capacity of irisflorentin to inhibit DC-induced allogeneic T-cell proliferation (Fig. 8). Consequently, irisflorentin modulated the activation of NF-κB and MAPK signal transduction pathways that participated in DC maturation by arresting IKK, JNK, and p38 activity.

Irisflorentin decreased the activation of IKK, NF-κB, and MAPK signaling pathways in LPS-stimulated mBM-DCs. mBM-DCs were pretreated with 20 or 40 μM irisflorentin. After 1 h of incubation, the cells were washed, followed by stimulation with LPS (100 ng/ml) for 40 min. Cells were lysed and phosphorylation levels (p) of IKKα/β, IkBα, JNK, ERK1/2, p38 MAPK, and Akt were analyzed by immunoblot analysis. One representative result from three independent experiments is shown. The expression of β-actin was used as an internal control. The relative fold in protein level was represented as the level in pretreated cells relative to that in irisflorentin-untreated, LPS-unstimulated controls. The data represent the mean ± SD (n = 3). A hash (#) shows significant differences between LPS-stimulated and unstimulated cells (#p < 0.001); an asterisk (*) shows significant differences between the LPS-stimulated control samples and irisflorentin-pretreated, LPS-stimulated samples (*p < 0.05, **p < 0.01).

IKK, JNK, and p38 MAPK pathways controlled allostimulatory ability of irisflorentin-pretreated, LPS-stimulated mBM-DCs. mBM-DCs were preincubated with the indicated inhibitors for 30 min and then treated with or without 40 μM irisflorentin. After 1 h of incubation, the cells were washed, followed by stimulation with LPS (100 ng/ml) for 20 h. Finally, the cells were washed and diluted with the prepared CFSE-labeled splenocytes in a ratio of 1:10 in culture plates for 3 days. The proliferation of T-cells was assessed by flow cytometry. The values of LPS-stimulated DCs served as control values in the calculation of percentage of proliferation. The data represent the mean ± SD (n = 3). A hash (#) shows significant differences between LPS-stimulated and unstimulated cells (#p < 0.001); an asterisk (*) indicates significant differences between the LPS-stimulated control samples and irisflorentin-pretreated (or inhibitors-preincubated/irisflorentin-pretreated), LPS-stimulated samples (**p < 0.01).
Irisflorentin Weakened Contact Hypersensitivity (CHS) Responses
We established an obstructive effect of irisflorentin on DC maturation, which implies that irisflorentin may prevent DC-mediated disorders. CHS is a simple in vivo test of cell-mediated immune response in which exposure of epidermal cells to exogenous haptens induces a delayed-type hypersensitive reaction that can be measured and quantified. Therefore, we carried out DNFB-induced CHS response as a model to test this hypothesis. Mice were sensitized by painting DNFB in the absence or presence of irisflorentin directly onto the abdomen. The CHS response to DNFB was then examined. The ears were obviously swollen in DNFB-sensitized, but not in DNFB plus irisflorentin-sensitized mice (100 μg, p < 0.01), whereas DMSO had no influence on DNFB-sensitized mice (Fig. 9), suggesting that irisflorentin inhibits the DC-mediated sensitization in CHS. These results indicate that irisflorentin has the potential to improve delayed-type hypersensitive disorders, such as allergic contact dermatitis.

Irisflorentin weakened the CHSR in mice. Mice were sensitized by painting DNFB with or without irisflorentin directly onto the abdomen. After 6 days, mice were challenged by painting on the backs of ears with DNFB. Ear thicknesses were measured 24 h later. CHS responses were showed by hematoxylin and eosin staining (A), and thickness of the challenged ear was recorded (original magnification 40×) (B). Mice that were not sensitized but were challenged with DNFB were provided as negative controls. One representative result from three independent experiments is shown. The data represent the mean ± SD (n = 3). A hash (#) shows significant differences between unsensitized and sensitized mice (#p < 0.001); an asterisk (*) shows significant differences between the DNFB-sensitized control samples and DNFB plus irisflorentin-sensitized samples (**p < 0.01).
Discussion
Our investigational data reveal that irisflorentin decreases the secretion of LPS-induced preinflammatory cytokines TNF-α, IL-6, and IL-12p70 by DCs, reduces LPS-induced expression of MHC class II, CD40, and CD86 molecules by DCs, and attenuates LPS-induced, DC-triggered allogeneic T-cell proliferation. These consequences support the claims of traditional Chinese medicine practitioners about the employment of herbs containing irisflorentin in the treatment of inflammatory-related diseases (36). To the best of our knowledge, this is the first report to demonstrate the immunosuppressive function of irisflorentin on DC maturation. Some studies have indicated that isoflavones, including genistein, daidzein (34), and glabridin (21), have special immunosuppressive effects on DCs. This study shows that irisflorentin is a new member on the list of isoflavones with these effects. The use of this isoflavone may provide a convenient means of controlling the immunomodulatory capacity of DCs.
TNF-α plays a central role in inducing production of IL-1, IL-6, platelet-derived growth factor, and transforming growth factor as well as adhesion molecules to mediate the inflammatory response. Furthermore, TNF-α also promotes DC survival and increases the production of reactive oxygen and nitrogen species by leukocytes (18,32). IL-6 has been implicated in a plethora of physiological functions, such as antibody production in activated B-cells, proliferation, differentiation, and migration of lymphocytes, control of Th1-associated cytokine expression and IL-2 receptor, and upregulation of acute phase proteins in the liver (18). Moreover, IL-6 has recently been reported to contribute in Th17 differentiation (4). IL-12 is essential for the proliferation, differentiation, and maintenance of Th1 that induces IFN-γ and IL-2 expression and CD8+ T-cell clonal expansion, function, and memory (8). Therefore, irisflorentin arrested the expression of TNF-α, IL-6, and IL-12 to inhibit T-cells and an inflammatory response. CD40 has a very crucial role in licensing DCs to prime naive CD8+ T-cell responses. Declining expression of CD40 molecules on DCs could destroy antigen-presenting capability, impair both primary and memory T-cell responses, diminish cytokine production, and abolish CD80, CD86, and CD70 expression (28).
In this research, we found that irisflorentin breaks multiple intracellular signaling pathways downstream of TLR4 in DCs (Fig. 10). The cytoplasmic protein, IκB, associates with NF-κB dimer, preventing their nuclear translocation. Starting of TLR4 activity by the LPS complex causes phosphorylation of IκB, followed by their degradation and the release of NF-κB, which is accordingly translocated into the nucleus. Activated NF-κB regulates proinflammatory-related gene expression, including cytokines, costimulatory molecules, and adhesion molecules as well as the maturation marker CD83 in DCs, and is upregulated when DCs mature (15). Irisflorentin decreased the activation of NF-κB by arresting the degradation of IκBα and the nuclear translocation of p65 in LPS-stimulated DCs. MAPK signaling pathways also play an essential role in DC maturation. In this study, we have confirmed that irisflorentin modifies LPS-induced activation of DCs, at least in part, through suppression of JNK and p38 MAPK pathways. DCs prolong the life span when activities of NF-κB and JNK/activator protein-1(AP-1) are in a balanced situation by the ligation of tumor necrosis factor receptor superfamily (TNFR-SF) members. A key role of NF-κB in DCs is to control the amount of JNK activity. When NF-κB activation lessens, TNFR-SF member-induced JNK activity is increased. Then DCs undergo JNK/AP-1-regulated apoptosis (22). Our results showed that JNK/AP-1 and NF-κB activities are attenuated simultaneously on irisflorentin-pretreated LPS-stimulated DCs. This induces TNFR-SF member-mediated survival of DCs. The p38 MAPK pathway positively regulates cytokine production and phenotype of mature DCs by the cAMP-response element-binding protein (CREB) and activation transcription factor 1 (ATF1) (1). Therefore, irisflorentin inhibits the activation of p38 MAPK-CREB/ATF1 and blocks DC maturation.
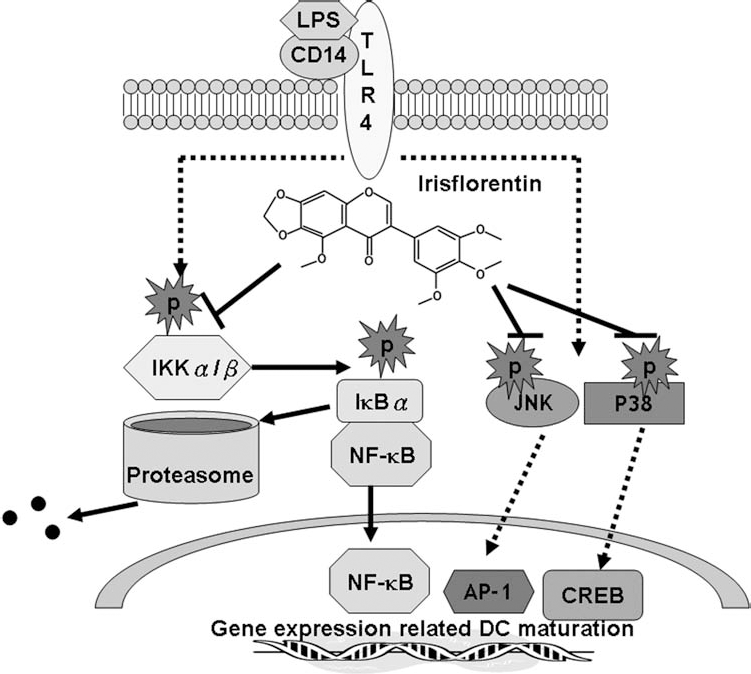
Possible mechanisms of immunomodulation of irisflorentin. Irisflorentin arrests the activity of mBM-DCs by blocking of IKK/NF-κB and JNK/p38 MAPK signaling. TLR-4, Toll-like receptor 4; AP-1, activator protein 1; CREB, cAMP response element-binding protein.
Recent advancements in manipulating DC–T-cell interactions for the treatment of human diseases hold great potential. Allergic contact hypersensitivity responses (CHSR) are demonstrated clinically as ACD, a general skin disease resulting from skin exposure to industrial or environment allergens. Langerhans cells (LCs, dermal DCs) have been manifested to act as primary antigen-presenting cells for the induction of CHSR in the sensitization phase (30). Therefore, our central hypothesis is that allergic CHSR should be preventable by arresting migration, maturation of LCs, and LC-induced T-cell activation (10,11). In this study, we showed that irisflorentin can improve CHSR by blocking DC function. In future work, we will investigate whether pretreatment of irisflorentin allows improvement in other inflammatory or autoimmunological diseases.
Footnotes
Acknowledgments
This work was supported by the Ministry of Science and Technology (Taiwan) (MOST 103–2314-B-039 -025), the Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence (MOHW104-TDU-B-212–113002), and China Medical University (DMR103–054). The authors declare no conflicts of interest.