
Abstract
Islet grafts can contribute to their own destruction via the elaboration of proinflammatory genes, many of which are transcriptionally regulated by nuclear factor κ-light-chain-enhancer of activated B-cells (NF-κB). Thus, NF-κB constitutes an enticing gene therapy candidate to improve the success of islet transplantation. To test this hypothesis in vivo, we blocked NF-κB in BALB/c (H2d) to C57/BL6 (H2b) mouse islet allografts by genetically engineering islets to express the NF-κB superrepressor, IκBα. Here we show by microarray and RTqPCR that islets exhibit an intrinsic early immediate proinflammatory response, with the most highly upregulated proinflammatory genes comprising the chemokines Cxcl1, Cxcl2, Cxcl10, and Ccl2; the cytokines Tnf-α and Il-6; and the adhesion molecule Icam1. Overexpression of IκBα inhibited the expression of these genes by 50–95% in islets and MIN6 β-cells in vitro, by inhibiting NF-κB-dependent gene transcription. Histological and RTqPCR analysis at postoperative day (POD) 10 revealed that IκBα-transduced islet allografts exhibited improved islet architecture and strong insulin-labeling with decreased Ccl2 and Il-6 mRNA levels compared to the GFP-transduced control grafts. Despite these protective effects, NF-κB-blocked islet allografts were promptly rejected in our MHC-mismatched mouse model. However, IκBα-expressing grafts did harbor localized “pockets” of Foxp3+ mononuclear cells not evident in the control grafts. This result suggested that the effect of the NF-κB blockade might synergize with regulatory T-cell-sparing rapamycin. Indeed, combining intragraft IκBα expression with low-dose rapamycin increased the mean survival time of islet allografts from 20 to 81 days, with 20% of the grafts surviving for greater than 100 days. In conclusion, rapamycin unmasks the protective potential of intragraft NF-κB blockade, which can, in some cases, permit permanent allograft survival without continuous systemic immunosuppression.
Keywords
Introduction
Transplantation of islets of Langerhans is a potential therapy for the treatment of type 1 diabetes (T1D), and may also benefit people with type 2 diabetes mellitus (T2DM), caused, in part, by β-cell insufficiency (31). However, inflammation remains one of the key barriers inhibiting successful islet transplantation. This is because inflammation links and exacerbates detrimental mechanisms of metabolic deregulation and islet cell death (mass loss), as well as innate and adaptive antigraft immunity (29,37).
It is this aberrant inflammatory response following transplantation that also renders islets active participants in their own destruction. Indeed, human islets express inflammatory factors upon isolation, culture, and transplantation (10). Consequently, transplanted islets have been shown to contribute to their apoptotic destruction via the induction of β-cell-toxic molecules, such as induced nitric oxide synthase (iNOS) (16), cyclooxygenase 2 (COX-2) (32), and activating transcription factor 3 (ATF3) (19); aid the recruitment of invading lymphocytes through the elaboration of chemokines [e.g., chemokine (C-X-C motif) ligand 10 (CXCL10), chemokine (C-C motif) ligand 2 (CCL2)] (8,13); and facilitate their own T cell-mediated killing through the expression of cytokines and death receptors, such as Fas/cluster of differentiation 95 (CD95) (9).
For these reasons, it is important to understand what molecular pathways within islet grafts contribute to inflammation. Gene expression (10), DNA-binding activity analysis (7), and pathway studies (1,7) have revealed nuclear factor κ-light-chain-enhancer of activated B-cells (NF-κB) as the central mediator of the aberrant inflammatory response in freshly isolated and cultured islets. Accordingly, the top-ranking genes associated with inflammation and apoptosis expressed during isolation and culture are NF-κB regulated (10). As a result, a NF-κB molecular signature in human islets prior to transplantation can predict graft failure (10). Therefore, NF-κB may constitute a promising therapeutic target to blunt the proinflammatory gene response to improve the success of islet transplantation.
Canonical NF-κB activation primarily predominates in human β-cells (7) and is strongly activated by tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in islets (16), potent inflammatory cytokines during allograft rejection. A critical step for NF-κB activation is phosphorylation and activation of the inhibitor of κB (IκB) kinase complex, which phosphorylates inhibitor of κB protein (IκBα). IκBα retains the NF-κB transcription factor in the cytoplasm, but upon phosphorylation, IκBα is targeted for ubiquination and degradation by the proteasome. This reveals the nuclear localization signal on the p65 NF-κB subunit, triggering its translocation to the nucleus where it transactivates target genes (20).
To explore whether the targeted blockade of NF-κB in islet allografts does constitute a mechanism to improve the success of islet transplantation, we examined the effect of inhibiting islet intrinsic NF-κB activation on islet allograft survival with or without additional immunosuppression. To achieve this, NF-κB activation was blocked by overexpressing the physiologic NF-κB inhibitor, IκBα (3). Our study demonstrates, in a robust preclinical model of allotransplantation, the benefits of graft-targeted NF-κB blockade. When NF-κB blockade was used alone, grafts showed improved architecture, reduced inflammation, and pockets of forkhead box P3-positive (Foxp3+) mononuclear cells at early time points; however, NF-κB blockade was unable to prolong islet allograft survival. In contrast, when coupled with low-dose rapamycin, some IκBα-expressing grafts showed permanent allograft survival. These findings present a way to avoid the contraindications associated with systemic immunosuppression.
Materials and Methods
Mice
C57BL/6 and BALB/c mice were purchased from the Animal BioResource Center (Sydney, Australia). All procedures and experiments conducted complied with institutional Animal Ethics Committee guidelines.
Mouse Islet Isolation
In brief, mouse islets were isolated by perfusion of the distended pancreas with 25 mg/ml of Collagenase I/II Blend Research Grade (Roche, Indianapolis, IN, USA). The tissue was then digested at 37°C for 13 min, washed in M199 + 10% bovine calf serum (BCS) (HyClone, Waltham, MA, USA), passed through a 425-μm sieve (US standard sieve series, A.S.T.E. E-11 specifications dual MFG, Co., Chicago, IL, USA), and the resultant tissue was separated on Ficoll-Paque Plus (GE Healthcare Australia Pty. Ltd., NSW, Australia). Islets were washed in M199 + 10% BCS and were then ready for further treatment.
Rodent Islet Isolation and Transplantation
Rodent islets were isolated from BALB/c (H2d) mice and 150–200 hand-counted islets transplanted under the kidney capsule of C57BL/6 (H-2b) mice (36). This strain combination represents a complete major histocompatibility complex (MHC) mismatch. Recipient mice were rendered diabetic by streptozotocin (Sigma-Aldrich, Sydney, Australia) injection (180 mg/dl, IP). Diabetes was determined as blood glucose levels ≥20 mM on two consecutive days and was measured via the use of a FreeStyleLite® glucometer and blood glucose test strips (Abbott Diabetes Care, Abbott Park, IL, USA). Blood was obtained by tail tipping. Islet viability was assessed using the LIVE/DEAD® Viability/Cytotoxicity Kit (Molecular Probes, Eugene, OR, USA) following the manufacturer's instructions. Rapamycin (LC laboratories, Woburn, MA, USA) was dissolved in vehicle solution (0.2% carboxylethyl cellulose, 0.25% polysorbate-80 in 0.9% NaCl; all Sigma-Aldrich) and injected IP on the day of transplantation and every day thereafter for 7 days.
Recombinant Adenovirus-Mediated Gene Transfer
Isolated BALB/c Islets (Australian BioResources, Sydney, Australia) and mouse insulinoma (MIN6) cells [sourced from authors of ref. (25)] were transduced with recombinant adenovirus (rAd.) to overexpress green fluorescent protein (GFP; rAd.GFP) or porcine IκBα (rAd. IκBα) (12) following the procedures we have established previously (16,17). For islet gene transduction, islets were inoculated with virus at the multiplicity of infection (MOI) of 10:1 and were incubated for 1.5 h at 37°C in 0.5 ml serum-free Roswell Park Memorial Institute (RPMI)-1640 medium (Gibco, Melbourne, Australia). Islets were then ready for further culture or transplantation. MIN6 β-cells were plated at a density of ~1 × 106/well in six-well tissue culture plates (Corning, Costar, Sydney, Australia) and were inoculated with virus at the predetermined optimal MOI of 100:1 in Dulbecco's modified Eagle's medium (DMEM; Gibco). After 1.5 h, cells were replenished with DMEM 10% fetal calf serum (FCS; Thermo Scientific, Melbourne, Australia) and cultured a further 24 h before use. The rAd.IκBα construct was obtained and maintained as described previously (12).
Determination of Transgene Expression
GFP expression was determined by fluorescent microscopy, and images were captured under a Zeiss inverted fluorescence microscope (Carl Zeiss Inc., Jena, Germany). Fluorescent-activated cell sorting (FACS) analysis was also used to assess transduction efficiency of cell lines following cell suspension in FACS buffer [1% bovine serum albumin (BSA); 1 mM ethylenediaminetetraacetic acid (EDTA) in 1× phosphate-buffered saline (PBS; all Sigma-Aldrich)]. To determine protein expression, primary islets or MIN6 β-cells were cultured for 24 h after gene transduction and lysed in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich); ~10 μg of total protein was resolved on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel (Sigma-Aldrich), and then the protein was transferred to a nitrocellulose membrane Immobilon-P® (Merck Millipore, Melbourne, Australia). Membranes were incubated with polyclonal anti-IκBα (Cell Signalling Technology, Danvers, MA, USA) antibody and followed by peroxidase-labeled secondary antibodies (Sigma-Aldrich). Signals were visualized using an ECL detection kit (GE Life Sciences, Sydney, Australia).
Determination of NF-κB Blockade
Nontransduced or rAd-infected islets (150–200 islets per treatment), or MIN6 β-cells (1 × 106 per six wells), were cultured for 24 h after viral transduction in RPMI 1640/DMEM 10% FCS and then stimulated with recombinant murine IL-1β (200 U/ml) (R&D Systems, Minneapolis, MN, USA) for 0–60 min. IκBα protein levels was assessed by immunoblot as described above. Reporter assays were carried out essentially as we have described (16). NF-κB activity experiments were conducted using MIN6 β-cells transfected with 0.3 μg of the NF-κB.Luc reporter (Promega, Sydney, Australia) and 0.25 μg cytomegalovirus (CMV).β-galactosidase (β-gal) (16). IκBα (12) or the empty pcDNA3.1 reporter was then added (0.3 μg), and each well was topped with 0.15 pcDNA3.1 to make 1 μg total DNA. Transfection was conducted using lipofectamine 2000 (Invitrogen, Melbourne, Australia) as per manufacturer's instructions. Following transfection, cells were stimulated with 200 U/ml of IL-1β. Luciferase activity was assayed in cell lysates harvested 8 h poststimulation using a luciferase assay kit (Promega). Results were normalized to β-gal or renilla activity (Galactostar; eBiosciences, San Diego, CA, USA) to give relative luciferase activity. Protein expression was determined by immunoblot, as described above. For some experiments, isolated islets were cultured overnight in the presence or absence of 200 U/ml each of TNF-α, IL-1β, and interferon-γ (IFN-γ) (R&D Systems) at 37°C in 0.5 ml of 10% FCS (Thermo Scientific, Melbourne, Australia) RPMI-1640 medium (Gibco).
Gene Expression Analysis by Microarray and Real-Time Quantitative PCR (RTqPCR)
Mouse islets were isolated and cultured from BALB/c donors as described above (23), and 150–200 islets per treatment were stimulated with 200 U/ml of mouserecombinant IL-1β (R&D Systems) for 1 h. Messenger RNA was extracted and subjected to microarray analysis as we described (23) using a custom microarray containing probe sets for 890 inflammatory genes. Prior to hybridization, the total RNA was amplified and labeled using the MessageAmp II aRNA Amplification Kit (Ambion, Austin, TX, USA). Amplified RNA quality was evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Samples were hybridized on a custom-designed “CustomArray-12K-Microarray” platform (CombiMatrix Diagnostics, Irvine, CA, USA). For each experiment, islets were isolated from 10 mice and were pooled. Each experiment was performed on four (BALB/c) independent islet preparations. The mean coefficient of variances (CVs) based on replicate features on the microarrays were ≤20%. For RTqPCR, the total RNA was extracted using the RNeasyPlus Mini Kit (Qiagen, Chadstone Centre, VIC, Australia) according to the manufacturer's instructions. Single-strand cDNA was produced using the Superscript III Reverse Transcriptase Kit (Invitrogen) according to the manufacturer's instructions. Primers were designed using Primer3 (http://primer3.wi.mit.edu/) with sequences obtained from Genbank and synthesized by Sigma Aldrich (Table 1). PCR reactions were performed on the LightCycler® 480 Real-Time PCR System [Rotor-Gene (Qiagen, Germantown, MD, USA)] using the FastStart SYBR Green Master Mix (Roche Diagnostics, Castle Hill, NSW, Australia). The real-time PCR conditions were initial denaturation, 95°C for 10 min, a three-step cycling condition for 40 cycles of 95°C for 15 s, 63°C for 30 s, and 72°C for 30 s. The threshold cycle (Ct) was set using the LightCycler 480 Software v1.5.0. Relative expression for each gene was analyzed using the comparative Ct method and was normalized to the expression of cyclophilin A. Fold change for genes was calculated following the 2ΔΔCT method comparing the relative expression between control and treatment groups (24).
Mouse Primers Used for RTqPCR Analysis, Designed Using Primer3 and With Sequences Obtained From Genbank and Synthesized by Sigma Aldrich

Cxcl2, chemokine (C-X-C motif) ligand 2; Ccl2, chemokine (C-C motif) ligand 2; Icam1, intracellular adhesion molecule 1; Tnf-α, tumor necrosis factor-α; Il-6, interleukin-6; Foxp3, forkhead box P3; Tgf-β, transforming growth factor-β; Ctla4, cytotoxic T-lymphocyteassociated antigen 4; Ifn-γ, interferon-γ; Cph, cyclophilin.
Immunohistochemistry
Tissues were fixed in 10% buffered formalin (Sigma-Aldrich), and paraffin sections were stained with hematoxylin-eosin (H&E) (Sigma-Aldrich). Paralleled sections were stained for insulin or Foxp3 using polyclonal anti-rabbit (Cell Signaling Technology) or anti-mouse/rat Foxp3 (eBioscience). Foxp3 detection required antigen retrieval with 10 mM citrate, pH 6, using a pressure cooker (Dako, Sydney, Australia). Secondary antibodies, anti-guinea pig and anti-rat (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), were used to detect insulin and Foxp3, respectively. The Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) was used to amplify the signal, and diaminobenzidine (DAB; Sigma-Aldrich) was used for visualization. Sections were counterstained with hematoxylin. CD4 or CD8 staining of paraffin sections was conducted at St. Vincent Hospital, Darlinghurst, Australia.
Statistical Analysis
For gene expression studies, statistical analysis was performed using the Students t test. For allograft survival, the day of rejection was plotted as Kaplan Meier curves and were analyzed using the Logrank test. Tests were conducted on Prism (v5) software (GraphPad Software, San Diego, CA, USA).
Results
Islets Exhibit an Intrinsic Early Immediate Proinflammatory Response
We begin our investigation by detailing the early immediate proinflammatory gene response of cytokineactivated islets (i.e., 1 h postactivation). This was to better understand the potential islet contribution to the antigraft proinflammatory response. For the experiment, primary mouse islets were stimulated with the proinflammatory cytokine, IL-1β, and their subsequent early immediate gene expression profile was analyzed by microarray analysis. Examination of the distribution of gene expression showed that 30 genes were highly upregulated (i.e., greater than twofold induction, p < 0.005) following IL-1β treatment (Fig. 1A). Examining this inflammatory transcriptome profile in detail, we found the most highly regulated transcripts to be of the chemokines Cxcl1 (induced ~19.4-fold), Ccl2 (approximately ninefold), and Cxcl10 (approximately fourfold). Also among the most highly regulated genes were transcripts for the cytokines Tnf-α (approximately fourfold) and Il-6 (~4.6-fold), as well as the adhesion molecules intracellular adhesion molecule 1 (Icam1; ~5.8-fold) and vascular cell adhesion molecule 1 (Vcam1; ~2.4-fold) (Fig. 1B). Interestingly, these factors are known to contribute to allograft rejection.
Consistent with our microarray data, RTqPCR analysis of mRNA expression in cytokine-activated primary islets demonstrated high expression of Cxcl1 (induced ~63-fold), Cxcl2 (~58-fold), Cxcl10 (~56-fold), Il-6 (~40-fold), Ccl2 (~24-fold), Icam1 (~20-fold), and Tnf-α (approximately sixfold) (Fig. 1C). The expression of these molecules was found to be β-cell intrinsic, as demonstrated by the analysis of IL-1β-stimulated MIN6 β-cells. In this case, Ccl2, Cxcl2, Cxcl1, and Icam1 were induced >1,000-fold, whereas Tnf-α was induced ~900-fold, Cxcl10 ~100-fold, and Il-6 approximately eightfold (Fig. 1D). These data demonstrate that islet grafts can make autonomous contributions to the ensuing posttransplant inflammatory response.
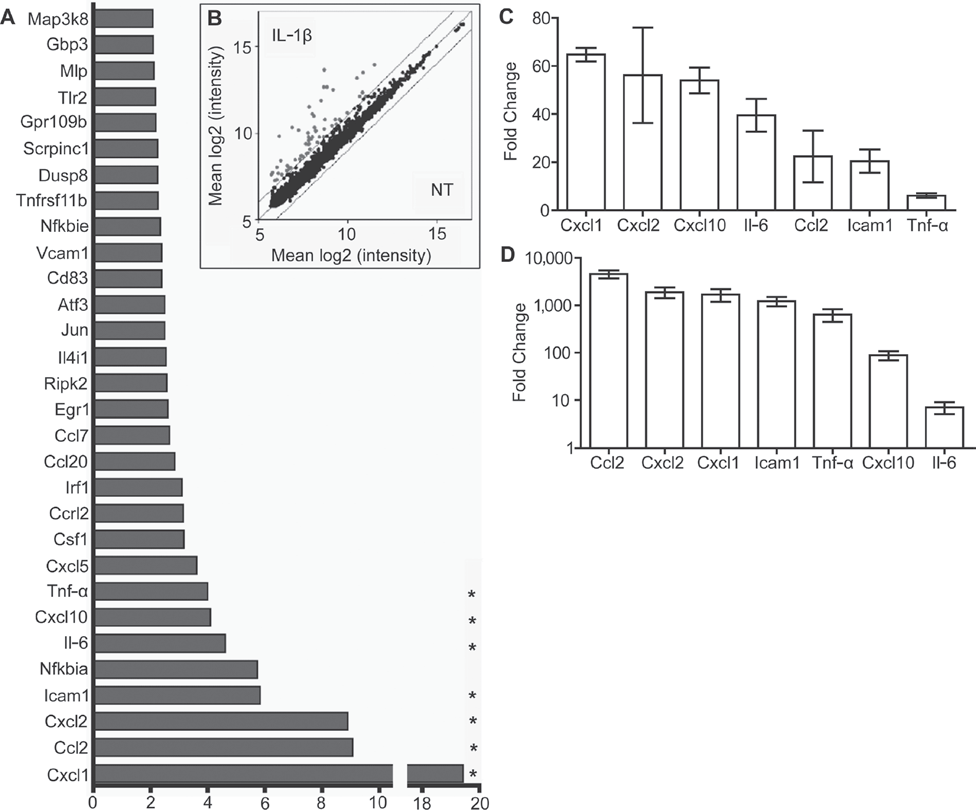
Islets exhibit a cell intrinsic early immediate proinflammatory gene response. (A) Thirty most highly upregulated genes in primary islets stimulated with interleukin (IL)-1β for 1 h. A value of p ≤ 0.005 for all genes induced greater than or equal to twofold. (B) Scatter plot analyses indicating the distribution of gene expression for IL-1β-treated primary islets 1 h poststimulation. Parallel diagonal lines indicate twofold cutoff. *Selected genes were confirmed by RTqPCR analysis in noninfected (C) primary islets and (D) mouse insulinoma (MIN6) β-cells. Cxcl1, chemokine (C-X-C motif) ligand 1; Ccl2, chemokine (C-C motif) ligand 2; Icam1, intracellular adhesion molecule 1; Tnf-α, tumor necrosis factor-α.
NF-κB Controls the Islets Early Immediate Proinflammatory Response
Many, if not all, of the early immediate genes we found to be expressed by cytokine-activated islets, contain response elements for the transcription factor NF-κB in their respective promoters. To determine whether NF-κB blockade would blunt the islets' early immediate proinflammatory response, we stimulated primary islets from BALB/c mice with 200 U/ml IL-1β, with or without IκBα, a physiologic inhibitor of NF-κB (3). Recombinant Ad.IκBα transduction was conducted at a multiplicity of infection (MOI) of 10:1 (Fig. 2A, B). Consequently, IκBα overexpressing primary islets exhibited a 50–95% reduction in the expression of a panel of inflammatory factors, including Cxcl1, Ccl2, Cxcl2, Icam1, Il-6, Cxcl10, and Tnf-α, compared to NI islets and GFP-overexpressing islets (Figs. 2C and 1C). We also observed a ≥90% inhibition of these same genes following stimulation of IκBα over-expressing MIN6 β-cells (MOI = 100:1) with 200 U/ml IL1-β, compared to noninfected (NI) and GFP-expressing MIN6 β-cells (Figs. 2D and 1D).

IκBα overexpression inhibits the early immediate proinflammatory response in islets. (A) Fluorescent microscopic image of a single islet 72 h posttransduction with recombinant adenovirus (rAd.) expressing green fluorescent protein (GFP), at a multiplicity of infection (MOI) of 10:1 (NI, noninfected). (B) Immunoblot analysis of inhibitor of κB (IκBα) expression in primary islets 24 h posttransduction with rAd.GFP (GFP) or rAd.IκBα (Ikba). GFP- or IκBα-transduced islets (C) or MIN6 β-cells (D) were treated with IL-1β for 4 h, and mRNA levels for proinflammatory factors were assessed by RTqPCR (*p < 0.05; **p < 0.01; 3-4 independent experiments).
IκBα Overexpression Blocks NF-κB Activation
Mechanistically, forced expression of IκBα in primary islets inhibited NF-κB signaling, at the level of IκBα degradation (Fig. 3). Islets overexpressing GFP and stimulated with 200 U/ml of TNF-α show a net degradation of IκBα at 15 min, which is indicative of NF-κB activation (16). This net degradation does not occur in IκBα-overexpressing islets (Fig. 3A). Overexpression of IκBα prevented the net degradation of IκBα in MIN6 β-cells when stimulated with IL-1β, and consistent with this result, this also inhibited NF-κB promoter activity (Fig. 3B, C). Together, these results support the concept that overexpression of IκBα prevents inflammatory gene expression by blocking NF-κB-dependent transcription. Second, they also indicate that NF-κB will need to be targeted either simultaneously or prior to the time of transplantation, given the rapid kinetic of the islet inflammatory response.

IκBα overexpression blocks NF-κB activation. (A) Immunoblot analysis of IκBα levels (arrows) in islets transduced with rAd.GFP (GFP) or rAd.IκBα (IκBα) and treated with 200 U/ml TNF-α for 0–30 min. (B) Immunoblot analysis of IκBα levels (arrows) in MIN6 β-cells stimulated with 200 U/ml IL-1β. (C) MIN6 β-cells cotransfected with a nuclear factor κ-light-chain-enhancer of activated B-cells (NF-κB).luc reporter and a cytomegalovirus (CMV).β-galactosidase (β-gal) construct with or without pcDNA3.1 encoding IκBα. Cells were then stimulated with 200 U/ml IL-1β or left untreated. Relative light units = Luciferase/β-gal (***p < 0.001; n = 3 independent experiments).
NF-κB Blockade by IκBα Overexpression in Islets Fails to Prolong Islet Allograft Survival
To test the effect of suppressing the islets' proinflammatory response via NF-κB blockade on allograft rejection, IκBα-transduced BALB/c (H2d) islets were transplanted under the kidney capsule of streptozotocin-induced diabetic C57BL/6 (H2b) mice (Fig. 4). Transduction at an MOI of 10:1 did not affect islet viability in vitro, as no difference in the amount of dead cells was observed 48 h posttransduction, when islets were costained with Calcein-AM and ethidium bromide homodimer-1 (data not shown). Transduced islets showed normal function in vivo (Fig. 4A), as GFP-transduced BALB/c (H-2d) islet allografts transplanted into streptozotocin-induced diabetic C57BL/6 (H-2b) mice easily restored metabolic control. Rejection rates were also no different between NI and control GFP-rAd-transduced grafts. Mice receiving (NI) (n = 11) or GFP-transduced (n = 9) islets rejected their grafts with a median survival time (MST) of 17 and 20 days, respectively (p = 0.56). Notably, grafts transduced with IκBα were also rapidly rejected (MST = 20 days; n = 10; p = 0.54) (Fig. 4B).

NF-κB blockade by IκBα fails to prolong islet allograft survival. (A) Blood glucose levels (BGL; mmol/L) of diabetic C57BL/6 (H2b) mice transplanted with allogenic (H2d) noninfected (NI; n = 4) or rAd.GFP-transduced (MOI = 10:1; n = 4) BALB/c islets; POD, postoperative day. (B) Kaplan-Meier survival data of BALB/c NI islet grafts [n = 11; median survival time (MST) = 17], rAd.GFP (GFP; n = 9; MST = 20; p = 0.56) or rAd.Iκbα (Iκbα; n = 10; MST = 20; p = 0.54) transduced grafts.
Improved Pathology of Islet Grafts Expressing IκBα
Analysis of IκBα- and GFP-overexpressing grafts was conducted at postoperative day (POD) 10 (Fig. 5). This included excising the graft-harboring kidney for the purpose of histology or RTqPCR. The objective of this experiment was to assess whether NF-κB blockade by IκBα rendered any early survival advantage over GFP-overexpressing grafts. Analysis of grafts by H&E and insulin staining revealed a striking difference between GFP-overexpressing grafts compared to IκBα-overexpressing grafts. IκBα-overexpressing grafts exhibited robust insulin staining and intact islet architecture at POD 10. In contrast, the control, GFP-overexpressing grafts exhibited fragmented insulin staining and less-defined islet architecture (Fig. 5A, B). Thus, IκBα-overexpressing grafts exhibited improved survival at this early time point. However, both grafts exhibited a similar level of immune infiltrate at POD 10 (Fig. 5A, B).

Histological analysis of NF-κB blocked islet allografts at POD 10. (A) Hematoxylin and eosin (H&E), (B) insulin (INS), (C) forkhead box P3 (Foxp3), *“region of Foxp3+ density” (D) cluster of differentiation 8 (CD8), or (E) CD4 staining of GFP control or IκBα-expressing grafts at POD 10. Scale bars: 50 μm (A–C) or 100 μm (D–E); n = 6. (F) Quantification of Foxp3+ cells [#per high-powered field (10×)].
Islet Grafts Expressing IκBα Show an Altered Inflammatory Profile
The improved pathology observed for IκBα-overexpressing grafts, compared to GFP-overexpressing grafts, may be attributed to a reduced NF-κB-dependant inflammatory profile in vivo. To test if this was the case, RTqPCR of grafts excised at POD 10 was conducted. Assessed genes included the most highly upregulated inflammatory genes identified by microarray analysis in Figure 1, such as Ccl2, Icam1, Il-6, Cxcl10, Tnf-α, Cxcl1, and Cxcl2. Consequently, significant inhibition for Ccl2 and Il-6 was detected at POD 10, with Icam1, Cxcl10, and Tnf-α being expressed at levels similar to that found in blockade had any effect on immune regulation at the graft site, we conducted histological analysis of the markers Foxp3, CD8, and CD4. RTqPCR analysis of regulatory-associated factors, Il-10, transforming growth factor-β (Tgf-β), interferon-γ (Ifn-γ), cytotoxic T-lymphocyteassociated antigen 4 (Ctla4), and Foxp3 was also conducted. We observed the presence of Foxp3+ cells in both GFP- and IκBα-expressing grafts at POD 10 (Fig. 5C). Interestingly, “pockets” of Foxp3+-rich areas were observed only for IκBα-expressing grafts. Despite the localized accumulation of Foxp3+ cells for IκBα-expressing grafts, no difference was noted in the overall frequency of Foxp3+ cells in this group compared with the GFP-expressing grafts (Fig. 5F). Furthermore, a subtle trend to increased levels of Il-10, Tgf-β, and Ctla4 that did not reach statistical significance was also observed for IκBα-expressing grafts versus controls (Fig. 6B). No significant change was observed in CD4 or CD8T cell levels upon anti-CD4, CD8 staining of grafts at POD 10 (Fig. 5D, E).
Low-Dose Rapamycin Synergizes with Intragraft NF-κB Blockade
We interpret our results to indicate that intragraft IκBα-mediated blockade of NF-κB had a positive effect upon graft pathology and local inflammation. But alone, the control GFP-overexpressing grafts at this time point (Fig. 6A). Cxcl1 and Cxcl2 were not detected at POD 10, which agrees with past observations showing that these chemokines are expressed only in the early immediate posttransplant time period. Overall, the data indicates that IκBα overexpression suppresses graft inflammation resulting in a reduced inflammatory milieu at POD 10 in vivo.
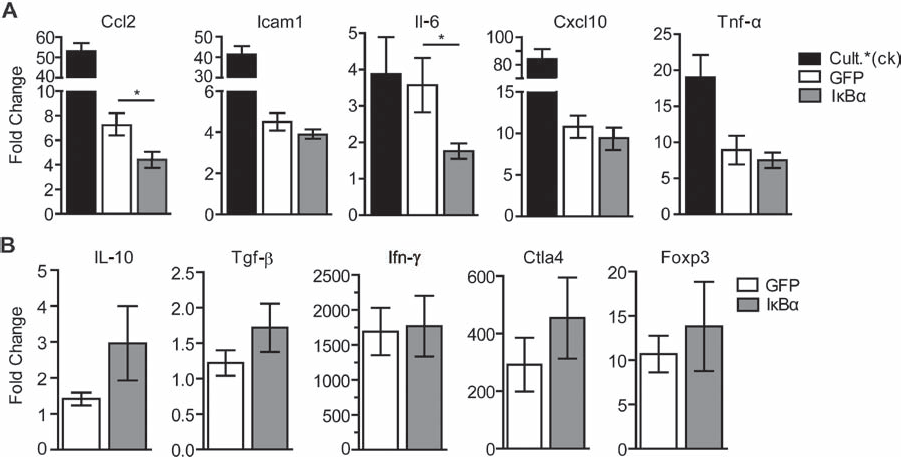
Assessment of inflammatory and regulatory factors in NF-κB blocked islet allografts at POD 10. rAd.GFP (GFP; open bar) or rAd.IκBα (IκBα; gray bar) overexpressing grafts were excised at POD 10 and subject to RTqPCR. Grafts were assessed for the topranking inflammatory factors found by microarray (Fig. 1) (A), or for regulatory-associated factors (B). Closed bars represent isolated islets cultured overnight and stimulated with 200 U/ml TNF-α, IL-1β, and interferon-γ (IFN-γ) as positive control [Cult. *(ck)]. All values were normalized against isolated islets cultured overnight without stimulation (*p < 0.05; n = 6). Tgf-β, transforming growth factor-b; Ctla4, cytotoxic T-lymphocyte-associated antigen 4.
Analysis of Regulatory Factors in IκBα-Targeted Grafts
One striking observation was the reduced level of Il-6 observed at POD 10. IL-6 promotes the differentiation of pathogenic T-helper 17 (Th17) cells and antagonizes CD4+ CD25+ Foxp3+ T-regulatory cell (Treg) generation (5). Therefore, a reduction in IL-6 levels may favor Treg generation in our model. To determine whether IκBα this was not sufficient to overcome the robust antigraft immune response in this stringent experimental model. In light of these observations, we considered that our approach might synergize with a Treg “sparing” immunosuppressive therapy (4,34). We decided to combine IκBα overexpression with rapamycin. First, we conducted a series of experiments with a scaled dose of rapamycin in a BALB/c (H2d) to C57BL/6 (H2b) islet transplant model, to find a dose regimen, which alone gave no significant benefit to graft survival (Fig. 7). Recipient mice given 1 mg/kg rapamycin on the day of transplantation, and everyday thereafter for 7 days, permanently accepted their graft (>100 days survival). Gradual reduction of this dose from 0.5, 0.2, to 0.1 mg/kg gave reduced MSTs of 43, 37, and 22 days, respectively. Therefore, a dose of 0.1 mg/kg gave no significant advantage in graft survival compared to the vehicle controls (MST = 19 days; p > 0.05) (Fig. 7A). When mice received 0.1 mg/kg rapamycin in combination with IκBα transduction, the MST of an islet allograft was increased from 17 to 81 days (p = 0.019) with ~20% of the grafts surviving long term (>100 days; p = 0.015 vs. GFP + 0.1 mg/kg rapamycin) (Fig. 7B).

Low-dose rapamycin synergizes with IκBα blockade to promote long-term islet allograft survival. (A) KaplanMeier survival data of BALB/c (H2d) noninfected (NI) islet grafts transplanted into C57BL/6 (H2b) streptozotocin-induced diabetic recipients. Recipient mice were given rapamycin (IP) at a dose of 1 (n = 3), 0.5 (n = 3), 0.2 (n = 6), 0.1 (n = 4), or 0 (n = 5) mg/kg, on the day of transplantation and every day thereafter for 7 days. (B) Kaplan-Meier survival data of BALB/c (H2d) IκBα-overexpressing islet grafts transplanted into C57BL/6 (H2b) streptozotocin-induced diabetic recipients, given 0.1 mg/kg regimen of rapamycin outlined in (A) (*p < 0.05 comparing IκBα + 0.1 mg/kg R to GFP; #p < 0.05 comparing IκBα + 0.1 mg/kg R to GFP + 0.1 mg/kg R; R, rapamycin; POD, postoperative day; IκBα + 0.1 mg/kg R. n = 5; GFP + 0.1 mg/kg R. n = 4; GFP n = 5; IκBα n = 5).
Discussion
The main finding of our study is that targeting graft-intrinsic NF-κB combined with an otherwise subtherapeutic regimen of rapamycin results in long-term allograft survival. This data is a marked development on previous studies targeting NF-κB, as it illustrates the potential benefit of graft-specific NF-κB blockade for clinical transplantation.
The rationale for targeting NF-κB in islets is strong. This is because human islets rapidly activate NF-κB during the islet isolation process (7,10), a transcription factor, which has been found to be the major regulator of rejection-relevant chemokines (22) and apoptosis in β-cells (16). Therefore, NF-κB activation in islets during isolation contributes to poor graft function and survival (10). An important milestone study by Giannoukakis et al. (14) first showed that blocking NF-κB in human islets by overexpressing IκBα, could protect islets from cytokine-induced cell death. Furthermore, key studies by Grey et al. (16) identified islet protective and anti-inflammatory effects of the A20 gene, which functions as a natural inhibitor of NF-κB activation in islets (15). That β-cells tightly regulate NF-κB with a physiologic A20-dependent feedback loop (23) also supports the concept that targeting NF-κB would be beneficial and also nontoxic for islets. Indeed, β-cell-specific overexpression of IκBα through a transgenic approach protected from streptozotocin-induced cell damage in vivo (11).
Following the stress of isolation, islets are also exposed to a toxic milieu once implanted. This mileu would include innate proinflammatory factors and also physiologic stresses driven by poor vascularization, resulting in conditions of local hypoxia and nutrient deprivation. Subsequently, islets exhibit poor function posttransplantation with apoptosis and necrosis claiming as much as 60% of the transplanted islet mass (6,18,26). Thus, targeting NF-κB during isolation or shortly after would act to dampen this islet-intrinsic proinflammatory response and would presumably promote optimal islet function when engrafted. Indeed, NF-κB blockade of islets prior to isolation, via systemic donor pretreatment with dehydroxymethylepoxyquinomicin (DHMEQ) or in situ transduction of IκBα inhibitor nemo-binding domain peptide, protects islets from apoptosis during islet isolation stress (28,33). In addition, targeting NF-κB in islet grafts via overexpression of A20, improved islet function in a syngeneic marginal mass transplant model, preserving β-cell mass and reducing cellular apoptosis (18). Further, Rink et al. (30) showed that conditional and specific suppression of NF-κB activity in β-cells improved graft function and survival in a syngeneic marginal mass transplantation model when transplanted into the clinically relevant site of the liver. Similar to those studies, we found that in our allograft model, IκBα-overexpressing grafts exhibited preserved architecture and robust insulin labeling. Thus, we conclude that some of the protective effects of IκBα for islet allografts relates to its ability to protect from toxic insults encountered in the early posttransplant period.
A further protective effect of IκBα overexpression related to its ability to dampen islet intrinsic inflammation, which as we show here, resulted in an altered inflammatory profile at the graft site. In one model of allograft rejection, immune danger, in the form of foreign MHC, graft debris, and inflammation would collectively act as potent alloreactive warning signals triggering the allograft rejection response. If subdued, the environment could favor a tolerant phenotype (2,21). Indeed, in our model, we observed reduced Ccl2 and Il-6 levels at POD 10. It is of interest that IL-6 promotes the differentiation of pathogenic Th17 cells and antagonizes CD4+ CD25+ Foxp3+ Treg generation (5). Therefore, reduced IL-6 levels may favor Treg generation. It is interesting to note that we did observe localized “pockets” of Foxp3+ mono-nuclear cells in IκBα-expressing grafts compared to the control grafts at POD 10. However, relative numbers of Foxp3+ cells in IκBα-expressing grafts versus the control grafts were not significantly different. We therefore hypothesized that IκBα overexpression might synergize with approaches that further favor Tregs, such as rapamycin (4,34). In support of this concept, we found that IκBα overexpression was permissive for a low dose, and short course of rapamycin, resulting in permanent islet allograft survival for some recipients.
NF-κB is a major regulator of immune cell function (20), making it an enticing target for therapeutic intervention in transplantation, and consistent with this, systemic blockade of NF-κB can prolong heart allograft survival (35). Further, targeting NF-κB selectively in lymphocytes with an IκBα transgene prolonged islet allograft survival (27). However, these approaches suffer the caveats associated with systemic immunosuppression. Indeed, in the context of islet transplantation, which potentially could benefit a pediatric population, avoidance of systemic immunosuppression would be preferable. We surmised that a reduction in graft-intrinsic inflammation might reduce the threshold of immune activation allowing for a reduction in the requirement for heavy immunosuppression. Here we demonstrate, in a robust preclinical model of islet allotransplantation, that islet intrinsic targeting of NF-κB reduces local inflammation, and when coupled with low-dose rapamycin, it allows for immunosuppression-free permanent allograft survival. Thus, our study demonstrates the benefits of graft-targeted NF-κB blockade as a way to avoid the contraindications associated with systemic immunosuppression.
Footnotes
Acknowledgments
We thank Dr. Andy McShea and Dr. Mariya W. Smith (Biology and Chemistry Section, CombiMatrix Corporation, Mukilteo, Washington) for technical and analytical assistance with microarray experiments. We thank Mr. E. Schmied (The Garvan Institute of Medical Research, Sydney, Australia) for providing valuable technical support and Professor S. Alexander (Centre for Kidney Research, The Children's Hospital at Westmead, Sydney, Australia) for the insightful discussion. N.W.Z. is supported by an Australian Postgraduate Award. S.T.G. is an ARC Future Fellow and an Honorary NHMRC Research Fellow. N.W.Z. designed the experiments, performed the experiments, interpreted the data, and cowrote the manuscript. B.M.T., S.N.W., D.L., J.E.V., and E.K.M. contributed to the establishment of protocols and provided intellectual input. S.T.G. designed the experiments, interpreted the data, and wrote the manuscript. The authors declare no conflict of interest.