
Abstract
Stress urinary incontinence (SUI) is a largely ousted but significant medical, social, and economic problem. Surveys suggest that nowadays approximately 10% of the male and 15% of the female population suffer from urinary incontinence at some stage in their lifetime. In women, two major etiologies contribute to SUI: degeneration of the urethral sphincter muscle controlling the closing mechanism of the bladder outflow and changes in lower pelvic organ position associated with degeneration of connective tissue or with mechanical stress, including obesity and load and tissue injury during pregnancy and delivery. In males, the reduction of the sphincter muscle function is sometimes due to surgical interventions as a consequence of prostate cancer treatment, benign prostate hyperplasia, or of neuropathical origin. Accordingly, for women and men different therapies were developed. In some cases, SUI can be treated by physical exercise, electrophysiological stimulation, and pharmacological interventions. If this fails to improve the situation, surgical interventions are required. In standard procedures, endoprostheses for mechanical support of the weakened tissue or mechanical valves for a bladder outflow control are implanted. In 20% of cases treated, repeat procedures are required as implants yield all sorts of side effects in time. Based on preclinical studies, the application of an advanced therapy medicinal product (ATMP) such as implantation of autologous cells may be a curative and long-lasting therapy for SUI. Cellular therapy could also be an option for men suffering from incontinence caused by injury of the nerves controlling the muscular sphincter system. Here we briefly report on human progenitor cells, especially on mesenchymal stromal cells (MSCs), their expansion and differentiation to smooth muscle or striated muscle cells in vitro, labeling of cells for in vivo imaging, concepts of improved, precise, yet gentle application of cells in muscle tissue, and monitoring of injected cells in situ.
Keywords
Introduction
Involuntary leakage of considerable amounts of urine after coughing, sudden movement, or heavy load is defined as stress urinary incontinence (SUI). The etiology of SUI is multifactorial and includes mechanical strain, aging, and chronic inflammation (22,45,57,66). In males, SUI may occur after prostate surgery (73,86). SUI yields a considerable impairment on the health and well-being of patients affected. This includes the quality of life, interaction with families and friends, expenses for auxiliaries, and may even promote symptoms of depression (45).
Current options for treatment include self-management and pelvic floor exercise (44), pharmacological intervention, and electrophysiological stimulation to improve muscular strength of the urinary sphincter and/or neuronal activation of the sphincter complex (32). When such regimens fail to improve the situation, surgical interventions, including injection of bulking agents, colposuspension, implantation of prostheses such as slings or tension-free vaginal tapes (TVT), or an artificial sphincter prosthesis, can be taken into consideration (45,53,86). Since conservative management for urinary incontinence in men after prostate surgery was often not satisfactory (10), artificial sphincter devices were developed. Recently, at least for some of these implant-based regimens, complications were reported (17,97). The complications include foreign body reactions to the material implanted, inflammation, and erosion (24). Moreover, slings, TVT, and comparable therapies only physically correct a displacement of the sphincter complex, or they apply mechanical pressure to the urethra to obstruct spontaneous urinary efflux; they do not treat the biological cause of incontinence: a neuronal or muscular deficiency.
The success of stem cell-based biological therapies in other fields, such as hematology–oncology or orthopedic surgery, raised hopes for patients and physicians that advanced therapy medical products (ATMP), such as cell-based therapies, could improve SUI. Preclinical studies to treat incontinence with progenitor cells or stem cells yielded promising results (11,27,33,54,59,60,63,76,80,89,90). This indicated that ATMP cell-based therapies potentially could improve the situation, at least for some SUI patients. Depending on the model employed, others reported no significant improvement of incontinence by cellular therapy in their preclinical studies (9). Experts agree that stem cell therapies are not yet state of the art in urology (27,37,54). Based on preclinical work completed, satisfactory clinical trials were initiated recently. In one study, treatment with autologous muscle-derived stem cells showed improvement in 4/8 female SUI patients after 1 year of follow-up, with one women even gaining continence (14). In another study, children (seven boys, one girl) were treated with autologous myoblast cells, and 88% showed improvement after 1 year as indicated by dryness for several hours during the daytime (51), and 5/7 boys were continent 4 years after myoblast implantation in another study (30). However, due to the follow-up time and growth of the children, it is unclear if all effects can be accounted for by the cell transplantation. Some clinical trials reported successes, but were retracted later (56,91,108). A clinical phase II study (HULPURO) for SUI treatment of women was approved recently (36). This study utilizes autologous adipose tissue-derived stromal cells (AT-MSCs) and was expected to be completed in 2014.
However, the debate on the optimal type of (stem) cell for treatment of SUI is not decided yet. In this regard, it is important to recall that the urinary sphincter is a contractile complex consisting of an inner muscular sheet generated by smooth muscle tissue and an outer Ω-shaped striated muscle (102) (Fig. 1). During development, these two different types of muscles are generated from different precursor cells. Development of striated muscles is activated by transcription factors located on chromosome 11 [myogenic factor 5 (Myf5), myogenic differentiation (MyoD)], and premyoblasts and myoblasts are differentiated from undetermined mesenchymal cells. In the adult, activation of satellite cells results in regeneration of striated muscle tissue (16,69,109). In contrast, mesenchymal stromal cells (MSCs) are precursors for development of the smooth muscle tissues in the inner organs and along the vasculature, and MSCs are the prime cells for regeneration of smooth muscle in the adult (74,83,84). We therefore summarize in this review some basic knowledge on the two most promising candidates for sphincter regeneration: muscle-derived satellite cells as precursor cells for repair of the rhabdosphincter, which is a striated muscle, and MSCs, derived from bone marrow or adipose tissue for general muscular regeneration and repair of the lissosphincter, which is a smooth muscle (Fig. 1).

Schematic overview on the male (left) and female (right) urinary sphincter complex. The urethra (gray tubing) is surrounded by a tubular muscle layer with longitudinal and circumferential muscle fibers built by smooth muscle cells and is called lissosphincter (dotted structure). The lissosphincter is enforced by an Ω-shaped muscle, built by striated muscle fibers, called the rhabdosphincter (striated Ω). The whole sphincter complex is stronger (thicker) in males compared to females but shorter. The details were published recently (102).
Myogenic Progenitor Cells for Sphincter Regeneration
As outlined above, in adults satellite cells are the precursor cells for striated muscle tissue (5,69). They are located between the basal lamina and the sarcolemma on muscle fibers (65). Upon stimulation by growth factors, hypoxia, or injury, quiescent satellite cells [integrin αC7+β1+, M-cadherin+, cluster of differentiation 34 (CD34+), paired box 7 (Pax7+)] are activated and start to proliferate and differentiate to become myoblast cells (Pax7low, MyoD+, Myf5+) (16,98,110), and myofibers [muscle-specific regulatory factor 4 positive (Mrf4+; Myf6+), myogenin+]. In a sense, this process recapitulates the gene expression sequence found in muscle tissue during embryonic development (Fig. 2). Activated satellite cells were shown to contribute to the regeneration of the urinary sphincter muscle (109). Thus, these cells were utilized in preclinical and clinical SUI studies (68,91). In addition, other myogenic precursor cells were defined in striated muscle tissue (23,67). Therefore, not only Pax7+ satellite cells (58) but also the interstitial precursor cells (PICs: PW1+, Pax7-, vimentin+) (67) or muscular pericytes (NG2+, ALP+) (23) may contribute to the regeneration of a striated muscle. In the context of this review, PICs seem to be an interesting type of cells for sphincter regeneration, as they are bipotent precursors. This means, at least upon stimulation in vitro, PICs can differentiate along the striated muscular lineage, and the smooth muscle lineage (67), and thereby serve to improve the two distinct muscular layers of the urinary sphincter complex, the rhabdosphincter and the lissosphincter (Fig. 1).

Stages of myogenic differentiation of muscular progenitor cells to generate striated muscular fibers. The principal muscular precursor cells are the satellite cells, which are derived during embryonic development from the somites (110). Upon activation by growth factors, hypoxia, or other stimuli, the resting satellite cells (G0 stage of cell cycle, CD34+, Pax7+) enter the cell cycle, proliferate (Myf5+, MyoD+), and differentiate along the striated muscle lineage (myogenin+) to generate multinucleated myofibers. Marker genes expressed during this differentiation pathways are indicated (98,110)
However, harvesting autologous satellite cells or other progenitor cells from a healthy striated muscle for repair of the urinary sphincter may bear some side effects. In addition, satellite cells or other muscular precursor cells are not very abundant in this type of tissue. Use of such cells therefore may require harvesting of even larger biopsies to isolate sufficient numbers of precursor cells for the treatment. Alternatively, the muscular precursors could be expanded in vitro to the amount of cells required for treatment. This strategy inherits other risks, including changes in the cellular phenotype or contaminations during in vitro expansion.
Therefore, investigators tried to generate multinucleated muscle fibers from other progenitor cells, which are available with less adverse effects. Possible candidates are multipotent MSCs abundantly available from bone marrow (BM-MSCs) (12,28,79,104), AT-MSCs (84,111,112), or other sources (18,19). Human MSCs have been characterized quite well, and they may act in two different ways upon injection in the urinary tract or in the sphincter complex specifically. They seem to differentiate to become muscle cells (29,33,34) and a wide differentiation potential of MSCs was reported (18,79). Differentiation of MSCs to smooth muscle cells was achieved by different methods: incubation of the AT-MSCs with epidermal growth factor (EGF-1), attachment to fibronectin, and high cell density cultures in the presence of corticosteroids generated smooth muscle cells (84). Other differentiation protocols employed transforming growth factor-β (TGF-β1) and bone morphogenetic protein-4 (BMP-4) for smooth muscle cell differentiation (103) or TGF-β1, angiotensin II, and blocking phospholipase C by sphingosylphosphorylcho-line (42). Differentiation of BM-MSCs to smooth muscle cells was achieved by stimulation of the cells with TGF-β1 (38), TGF-β1, and ascorbic acid (74) or TGF-β1 and PDGF-BB in three-dimensional scaffolds or alone (85,94). Although this list of procedures of induction of differentiation of MSCs to smooth muscle cells is far from being complete, it seems that TGF-β1-dependent SMAD2/3-signaling, and PDGF- or EGF-dependent MAP-kinase and STAT signaling seem to be important for this type of MSC differentiation (Fig. 3A). Interestingly, blocking the MAP-kinases also yielded induction of smooth muscle marker genes (92). However, two more or less distinct phenotypes or stages were described for vascular smooth muscle cells (Fig. 3B) (112). Therefore, differentiation of smooth muscle cells from MSCs may also yield distinct cells resembling either the synthetic or the contractile phenotype of smooth muscle cells. This two-sided phenotype of smooth muscle cells may account for seemingly conflicting data reported with respect to induction of expression of myogenic marker genes in MSCs after myogenic differentiation.
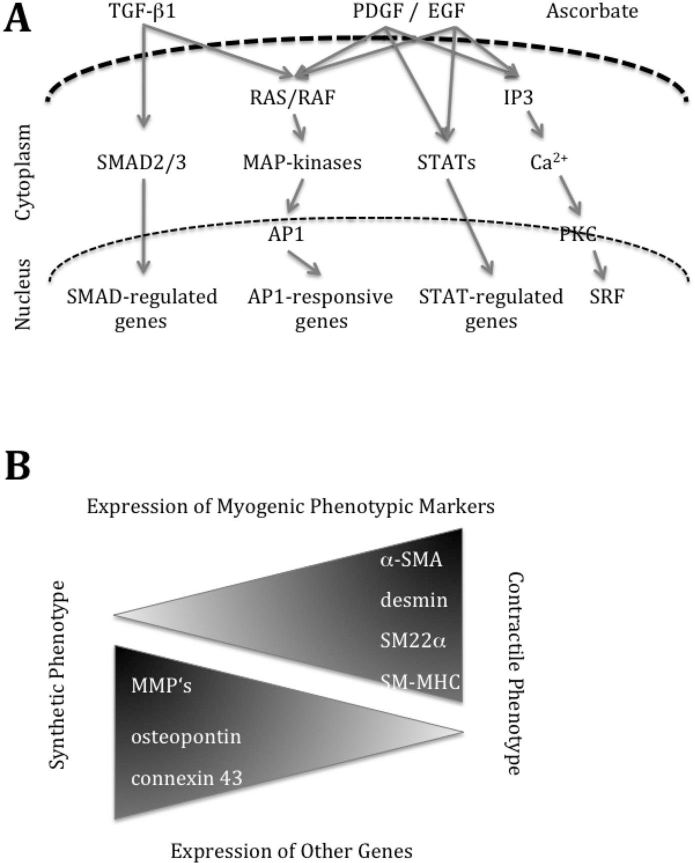
Paths of smooth muscle cell differentiation. (A) Simplified TGF-β- and PDGF- or EGF-dependent cytoplasmic signaling pathways. A comparative search of the literature reporting on generation of smooth muscle cells from MSCs indicates that most investigations utilized TGF-β1 or TGF-β1 plus PDGF for induction of myogenic marker genes (75). Abbreviations used: AP1, activator protein 1; IP3, inositol 1,4,5 triphosphate; MAP-kinase, mitogen-activated kinase; PKG, protein kinase G; RAS/RAF, Rous sarcoma/rapidly accelerated fibrosarcoma; STAT, signal transducer and activator of transcription. (B) Changes in gene expression in smooth muscle cells between the synthetic and proliferative stage (left) versus their contractile stage (right). During proliferation of smooth muscle cells, matrix metalloproteinases (MMPs), which are involved in tissue degradation, for example, during wound healing, osteopontin (synonyms: secreted phospoprotein 1, bone sialoprotein 1), a cytokine-regulating proliferation of many cells and connexin-43, a component of gap junctions and therefore important for cell–cell interaction, are expressed at higher levels. In contrast, the virtually resting contractile smooth muscle cells express the factors required for contraction including α-smooth muscle actin (α-SMA), desmin, a protein of the cytoskeleton important for structural and mechanical integrity of the cell, SM22α (alias transgelin), an actin-binding factor involved in regulation of muscular contraction, and smooth muscle myosin heavy chain (SM-MHC). More details were published recently (83).
MSCs were also differentiated along the striated muscular line, and elevated expression of MyoD, Myf5, myogenin, and myosin was shown after differentiation (25,35). These studies employed either recombinant technologies to express a myogenic stimulus in MSCs (25,39,40), mutagenic drugs (101), or xenobiotic serum (35). These methods are not acceptable for production of therapeutic cells. However, these studies help us to explore the myogenic differentiation pathways of MSCs (Fig. 4) and thus facilitate the design of future SUI therapies.
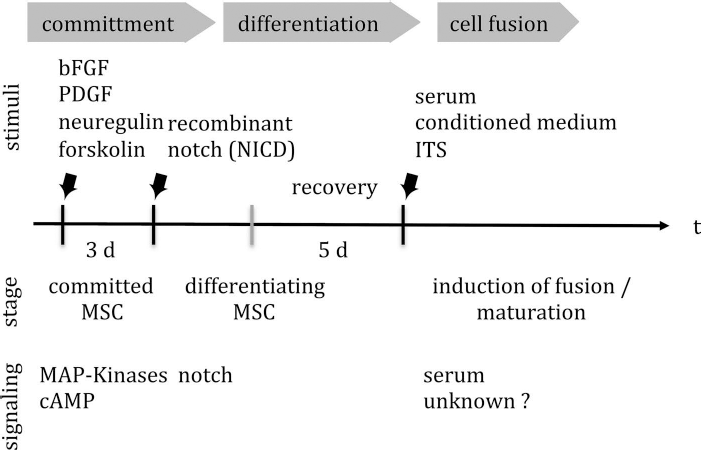
Induction of myogenic differentiation of MSCs to express markers of striated muscle cells. For myogenic differentiation MSCs pass through stages of commitment induced for instance by cytokines (bFGF, PDGF, neuregulin) in concert with additional stimuli (forskolin). After initial myogenic commitment, a transient Notch signaling is important to facilitate further differentiation of MSCs. By incubation of the MSCs in serum [xenobiotic serum, MSC-conditioned culture supernatant, ITS (insulin–transferin, selenium mix), possibly also by corticosteroids], generation of multinucleated myofibers was observed (25).
A general problem of the studies mentioned here is the fact that different protocols and methods were used for expansion, differentiation, quality measures, and analyses of differentiation of the cells. Therefore, the efficacies of these differentiation studies cannot be compared. Moreover, to the best of the knowledge of the authors, at present a protocol for efficient production of smooth muscle cells from human MSCs or striated muscle cells from MSCs under GMP-compliant conditions is not yet established.
On the other hand, for treatment of SUI with autologous MSCs, myogenic differentiation may not be necessary, and MSCs may be applied clinically without complex in vitro cell culture procedures. MSCs produce paracrine growth factors, and therefore were recently termed “a drugstore” for regenerative medicine (13). MSCs are known to express in vitro and in vivo cytokines that facilitate the regeneration of striated muscle [such as hepatocyte growth factor (HGF), insulin-like growth factor (IGF-1), IGF-2, and basic fibroblast growth factor (bFGF)] (6,21), smooth muscle [such as TGF-β1, platelet-derived growth factor (PDGF), bFGF)] (43), or cardiac muscle (26,49,82), as well as neuroregenerative factors [such as granulocyte colony-stimulating factor (G-CSF), vascular endothelial growth factor (VEGF), glial-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF)] (15,46,93). MSCs also promote the regeneration of vasculature (7,70). Therefore, MSCs act locally by production of regenerative factors and do not need to necessarily undergo differentiation to the mature effector cells such as smooth muscle cells or myofibers.
A direct application of bulk MSCs bears another specific risk: MSCs were shown to contain distinct subsets of cells that differ significantly in their phenotype (4,72,84). In addition, MSCs isolated from bone marrow or adipose tissue were shown to differentiate in vitro along the osteogenic lineage without addition of morphogenic factors and by only slowing mitosis through corticosteriods (28,79,112). This osteogenic differentiation of MSCs facilitates the generation of a stiff, calcified extracellular matrix. Such ossifications are possible side effects after application of bone marrow-derived cells, and this observed upon injection of bulk BM-MSCs in a healthy and infarcted heart muscle (8). This spontaneous ossification seems to be significantly less prominent in MSCs derived from human term placenta (78,96). Therefore, selection of the optimal source of autologous MSCs, bone marrow versus adipose tissue (or possibly for women postpartum, the maternal part of placenta), and determination of a suitable subset of MSCs may enable us in the future to apply autologous MSCs directly in vivo for the treatment of SUI. However, at present it is not known if the numbers of even distinct subsets of MSCs are sufficient to yield a measurable benefit to the SUI patients. We therefore continue to further improve current protocols for isolation, production, and application of MSCs and include in vitro cell culture to generate a sufficient number of cells for therapy. This requires, of course, standard operation procedures fully compliant with the good manufacturing procedures (GMP).
Gmp-Compliant Production of Cells for Clinical Purposes
Success in transfusion of blood and transplantation of tissues paved the way to develop cell injection or cell transplantation methods for treatment of human patients. Routine cell culture procedures to expand mammalian cells under sterile conditions were developed in the 1940s when sterile cell culture hoods and antibiotics were at hand. This raised hope that human cells could become a medication eventually. However, cell culture procedures meeting the current regulations set by the authorities (e.g., EMA in the EU; FDA in the US) require quite different protocols for isolation, characterization, and expansion of cells destined for ATMP therapy. In addition, quality measures to ensure phenotype, purity, and potency, as well as safety and efficacy, are rather strict (41). In this context, we can only briefly summarize some of the relevant aspects of GMP-compliant production of therapeutic cells.
All steps have to be performed under strictly contamination-free conditions, as antibiotics to control microbial contaminations are not allowed during processing or expansion of therapeutic cells at all. Second, prior to the application, complete sterility of the cells has to be proven. Third, all media needed for washing, expanding, harvesting, and packaging, transportation, or storage of the cells have to be exactly defined, and xenobiotic components, such as bovine serum or xenobiotic growth factors, are generally not acceptable. Therefore, specific cell culture media were developed for expansion of human therapeutic cells. In such media, human blood products, such as platelet extract, plasma, or serum, are blended to replace the standard cell culture components (fetal bovine serum, recombinant or xenobiotic growth factors, etc.) (31). Fourth, for referencing, backup samples have to be stored for quite some time, and fifth, each step from harvest of the cells from the donor to their implantation in the patient has to be described in standard operation procedures (SOP) and recorded in detail. Therefore, production of therapeutic cells by steps that include in vitro manipulations (for instance cell culture, growth on scaffolds, tissue engineering) is very labor-intensive, time-consuming, and therefore expensive. However, for many clinical applications, other options are not at hand, thus requiring the complex production of therapeutic cells in a laboratory.
Labeling Progenitor Cells for their Monitoring in Vivo
Fluorescent dyes, such as PKH26, PKH2, and PKH67 (Sigma-Aldrich, St. Louis, MO, USA), lipophilic carbocyanines (DiI, DiD, DiO, DiR, and the like; Molecular Probes, Life Technologies, Grand Island, NY, USA), or fluorescent nanocrystals (Molecular Probes) are suitable to label cells prior to their injection in live animals. The labeled cells can be located in tissue samples harvested after the desired period of incubation by simple fluorescence microscopy (Fig. 5). The labeling protocols for PKH dyes, lipophilic carbocyanines, or nanocrystals are rather simple, straightforward, and do not require recombinant technology nor a gene technology safety environment in the laboratory or animal facilities. Thus, the adherent cells or cells in suspension are incubated for a few hours (typically 1-24 h) in normal media enriched with the respective labeling components. Then the surplus of dye is removed by washing steps. No further equipment or facilities are needed. Therefore, such labels are a good option for a first evaluation in the context of preclinical cell injections; however, the fluorescent dyes are diluted with each cell division, as the dye is distributed to both daughter cells. Hence, long-term observations of proliferating cells (i.e., in vitro more than a few cell cycles, in vivo more than about 6 months) are probably impossible with this technique. For example, muscle cells, labeled by incorporation of a thymidine analog such as 5-ethynyl-2′-deoxyuridine (EdU) in the DNA, could be followed in urethral tissues only up to 3 months (2). Long-term labels are achieved best by recombinant technology. This however seems not (yet) acceptable in a clinical context. We therefore discuss here an option, which is at least fully compliant with a preclinical use of the cells: One may load the cells prior to injection with ferromagnetic particles to detect them in live animals by magnetic resonance imaging (MRI).
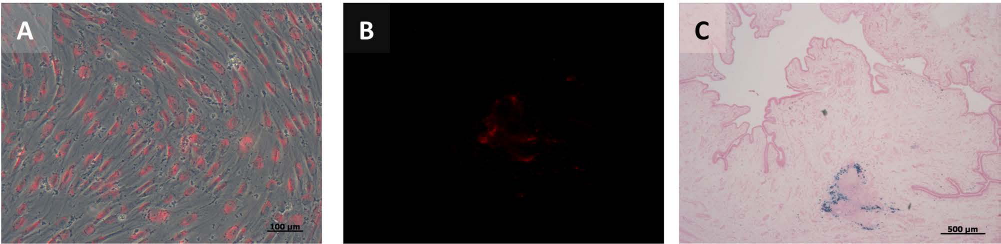
Labeling and visualization of cells in preclinical studies. In vitro labeling of adherent human adipose tissue-derived mesenchymal stromal cells resulted in homogenous staining after 4 h of PKH26 incubation (A). Detection of PKH26-labeled MSCs was demonstrated in situ through red fluorescence (B), and visualization of SPIO particles (Resovist®) in situ was performed via Prussian blue staining (C). Tissue sections from minipigs after 3 weeks of injection (B, C) are shown in 2.5x magnification.
Superparamagnetic iron oxide (SPIO) nanoparticles (e.g., ferucarbotran) can be used to label, for instance, cultured MSCs or other cells and to detect the injected cells in vivo by MRI. For some problems, anatomical data (location, migration, tissue distribution, density, etc.) in live animals must be linked with histological data (viability, local inflammation, phagocytosis of labeled cells, etc.) in tissue samples. In such cases, the cells can be tagged by a combination of fluorescent labels, such as SPIO nanoparticles plus a fluorescent dye (Fig. 5) (87). To this end, a good staining can be achieved when adherent cells are incubated overnight with the ferucarbotran nanoparticles. Then, after removing the free nanoparticles by flushing the cell with medium, the cells can be stained with the fluorescent label for the desired period of time. The labeled cells can then be detached and prepared for injection in preclinical in vivo models. For cells grown in suspension, this protocol may need some adjustments, but should basically work as well.
In preclinical models of cellular therapy, dual-labeled cells (fluorescence plus SPIO particles) can be detected after incubation for different lengths of time. Histological analyses of samples from the injection sites revealed that the cellular depots generated by simple injection of the labeled cells in a muscle remained stable (Fig. 5). Moreover, in our recent experiments, PKH26-labeled MSCs and SPIO particles were detected together in the urinary sphincter muscle by a combination of fluorescence microscopy and iron staining techniques up to 6 months after injection. We therefore conclude that cells double labeled with fluorescent dyes and SPIO nanoparticles can be tracked in vivo not only for a few days, but depending on i) the proliferation and migration activities of the cells applied, ii) the labeling technique applied, and iii) the tissue, for several weeks and even months.
In addition to the MRI labeling techniques described above, a different method for tracking cells in vivo is available: positron emission tomography (PET). PET is a quantitative imaging method that detects and quantifies small amounts of positron-emitter-labeled biomolecules. Direct cell labeling approaches of PET are based on commonly used radioactive compounds, for example, [18F] fluordeoxyglucose ([18F]FDG) or [64Cu]pyruvaldehyd-bis-(N4-nethylthiosemicarbazon) ([64Cu]PTSM). PTSM is a lipohilic redox-active carrier that delivers the radioactive [64Cu2+] into the cell (Fig. 6A). Subsequently, the reduction of Cu2+ to Cu+ in the cytoplasm opens the PTSM ring, and the free [64Cu+] is bound to cytoplasm proteins (1).

Detection of cells by positron emission tomography. (A) Schematic mechanism of [64Cu]PTSM labeling in cells. Reduction of [64Cu2+] to [64Cu+] in the cytoplasm and disposal of the empty PTSM from the cell leading to trapping of [64Cu+] within the cell. (B) Detection of transduced cells with HSV1-TK using [18F]FHBG for PET imaging. [18F]FHBG is phosphorylated by kinase activity and thus cannot diffuse out of the cell.
However, a main limiting factor for a longitudinal detection of directly labeled cells is the half-life time of such isotopes used for in vivo PET applications. Owing to the radioactive decay, the labeled cells can only be monitored for a certain amount of time. Other limiting factors of this method are the reduction of the detected signals due to an ongoing proliferation of the cells (55,77) and in addition the high efflux and the poor intracellular stability of the isotope within the cells (55). In contrast, labeling MSCs directly with [18F]FDG maintains their cellular properties, which is a benefit of this method (106). Recently clinical studies were performed and [18F] FDG-labeled human stem cells were injected intracoronarily in patients to monitor the effects, homing, and tissue distribution patterns (52). Performing [64Cu] labeling of monkey stem cells corroborated the feasibility of this method for in vitro studies (47).
Indirect approaches, where the stem cells are labeled not directly with a PET isotope, are based on reporter genes that are used to detect the stem cells for a longer time period (Fig. 6B). A generally used reporter gene is the herpes simplex virus1-thymidine kinase (HSV1-TK), which phosphorylates thymidine analogons. The PET tracer 9-(4-[18F]fluoro-3-hydroxymethylbutyl)guanine ([18F]FHBG) is such an analogon and can therefore be used to image those reporter genes. To detect the stem cells with such tracers, the cells need to be transduced with the HSV1-TK virus. After injection of the transduced cells, followed by an [18F] FHBG injection, the [18F]FHBG will be phosphorylated through the HSV-TK, resulting in a trapping of transduced cells. A technical advantage of this approach is the finding that not transfected cells generate a rather low background signal [18F]FHBG (107). However, a disadvantage of this approach is the fact that immune reactions can be provoked by recombinant cells.
Several studies using transduced cells are already published. For imaging tumor stroma and other tissues in mice, transduction of human MSCs with the HSV1-TK virus was employed (48). Others performed a proof-of-concept study after intramyocardial administration of the HSV1-sr39tk protein, and the labeled cells were detected using a clinical PET/computer tomography (CT) scanner (105). However, the possibility to use labeled stem cells to promote injury healing was first shown by Lee and colleagues. For this purpose, they transduced adipose-derived stem cells and monitored them in vivo by using PET and the tracer [18F]FHBG (61). Another approach showed the feasibility of labeling stem cells for in vivo bioluminescence and PET imaging (64). Thereby human stem cells were transduced by a triple-fusion reporter system based on the lentiviral vector fluc-mrfp-ttk, transplanted in mice and followed up by bioluminescence, computed tomography, and PET imaging.
These studies clearly demonstrated that stem cells can be labeled and tracked in vivo for a short time frame using the direct approaches based on [18F]FDG or [64Cu]PTSM and for a longer time frame by transducing the stem cells and using, for example, [18F]FHBG for PET imaging.
Injection of Cells in Preclinical Models
Labeled MSCs have been applied in different studies to explore their regenerative potential in vivo (14,36,70,71), and human MSCs have been utilized in xenogenic transplantation models of different diseases (20,29,62,81,88,95). In our current investigations, human MSCs are injected in porcine muscle tissue through a cystoscopic needle under sight, and 1.2-2.0 × 106 MSCs are injected into the rhabdosphincter in four depots with 250 μl of cell suspension each. For a better disposition of the PKH26-/Fe-labeled cells, the needle remained in the injection side for an additional 10 s (Fig. 5). However, depending on the tissue to be treated by application of regenerative cells, the strategies for application of the cells will differ. There is evidence that MSCs have the capability not only to regenerate defects in certain tissues by themselves (3,50,99,100) but also to facilitate the regeneration of various tissues by production of factors that activate resident stem cells, support vascularization, and promote infiltration of regenerative cells (13,71).
Depending on the label utilized to stain the cells prior to injection, migration and survival of the cells applied can be monitored in animals in vivo. However, some label may be spread from the originally stained cells to cells and tissue in close vicinity. This has been observed especially with PKH types of dyes. Moreover, label released from apoptotic or dead cells may be taken up by phagocytic cells and transported along lymphoid drainage or blood vessels. Combinations of “passive labels” (e.g., fluorescent dye, SPIO, etc.) and “live labels” (such as GFP expression, metabolic labels, etc.), and combinations of in vivo and ex vivo detection methods will facilitate the precise evaluation of the outcome.
Summary and Outlook
Investigation of mechanisms of tissue regeneration by injection of MSCs or other regeneration-competent cells is required, especially when vascularized and motile tissues, such as the urinary sphincter muscle, are targeted by cells. For in vitro purposes and preclinical in vivo studies, a plethora of dyes, nanoparticles, and enzymatic protocols allow staining or marking of virtually any cell for any imaging technique and for follow-up of cells, even in live animals. The principle problem lies with how to trace injected or implanted cells in human patients that allow appropriate in vivo follow-up. Needle biopsies could be theoretically used but do carry with them a significant morbidity, are uncomfortable, and large amounts of material cannot be removed because of the potential for damaging the urethral sphincter mechanism. Clearly, we need to have a more robust manner in which implanted cells can be studied, for instance, using fluorescent dye, as has been described with tumor-targeted fluorescent dyes used in tumor staging (98). Future developments will be necessary to allow us to effectively study implanted cells and identify the consequent function that develops, their location, and potentially, if possible, whether they become adequately innervated to provide a functional contribution to the urethral sphincter mechanism.
Footnotes
Acknowledgments
The authors thank Chaim Goziga for help in preparation of the illustrations and figures. Our own work mentioned in this review briefly was supported in part by grants from the German Research Council (DFG: KFO273), the Federal Ministry of Education and Research (BMBF: ReGiNA projects), the COST program of the EU (BM1209), and by institutional funds. The authors declare no conflicts of interest.