
Abstract
Manipulation of regulatory T cell (Treg) migration by islet expression of the chemokine CCL22 prevents diabetes in NOD mice and delays recurrent autoimmunity in syngeneic islet transplants. We sought to determine whether attracting Tregs with CCL22 also prevents islet allograft rejection. Isolated Bl/6 mouse islets were transduced overnight with adenovirus expressing CCL22 (Ad-CCL22) downstream of the CMV promoter. Islets were transplanted under the renal capsule of Balb/c recipients made diabetic by streptozotocin. To assess immunologic tolerance, graft-bearing kidneys from recipients of CCL22-expressing islet grafts were removed, and mice received a second transplant of naive islets from the same donor strain or third-party islets into the contralateral kidney. Adenoviral expression of CCL22 conferred prolonged protection of islet allografts in MHC-mismatched, diabetic recipients, maintaining normoglycemia in 75% of recipients for at least 80 days. Increased frequency of Treg cells was observed in islet grafts transduced with Ad-CCL22 compared with untreated grafts. Normoglycemic recipients of CCL22-expressing islet grafts showed complete absence of antidonor antibodies and no lymphocyte proliferation after exposure to donor splenocytes. After removal of the primary graft at day 80, mice that received a second transplant with untreated islets from the same donor strain did not reject the grafts, suggesting the development of tolerance. Expression of CCL22 recruits Treg cells to transplanted islets, prevents activation of alloreactive T-cells and islet allograft failure and induces alloantigen-specific tolerance. Manipulation of Treg cells by CCL22 in transplanted islets may be a novel therapeutic strategy for diabetes.
Introduction
Islet transplantation has emerged as a promising therapy for type 1 diabetes (25,30); however, most islet transplant recipients return to insulin injections within a few years posttransplantation (29). Numerous strategies in mice and humans have been tested and have shown improvement in short-term outcomes, but still have limitations. Allograft rejection remains a major hurdle in islet transplantation, given that cadaveric organ donors are not matched with the recipient for HLA type and that each patient typically receives islets from two or more donors.
The destruction of β-cells in islet allografts involves both CD4+ and CD8+ T cells, dendritic cells, and macrophages. Regulatory T cells (Tregs), a subpopulation of CD4+ T cells whose development and function depend on the transcription factor FoxP3, have attracted much attention because they are primary mediators of peripheral self-tolerance and immune homeostasis (12). There is a large body of literature in allotransplantation that shows correlation of islet protection with increased presence of Tregs (5,8,9,12,28).
We recently demonstrated that production of CCL22 in islets after intrapancreatic duct injection of double-stranded adeno-associated virus serotype 8 (dsAAV8) encoding CCL22 recruits endogenous Tregs, which have high constitutive expression of the receptor CCR4 (1,7,19), to the islet and confers protection from autoimmune diabetes in nonobese diabetic (NOD) mice (21). In addition, adenoviral expression of CCL22 in syngeneic islet transplants in diabetic NOD recipients prevents β-cell destruction by autoreactive T cells, delaying recurrence of diabetes (21). In the present study, we assessed the capacity of CCL22 to protect transplanted islets in the context of alloimmunity.
Materials and Methods
Viral Vector Production and Amplification
The adenoviral vector Ad-CCL22 was cloned as previously described (21). Briefly, a CCL22 cDNA was cloned in the pShuttle+/+ plasmid (ABM Good, Vancouver, BC, Canada). The adenovirus was constructed by cotransfection of HEK 293 cells (ATCC, Manassas, VA, USA) with the CCL22 vector and the adenovirus helper plasmid (ABM Good) and purified with Vivapure Adenopack 100 Kit (VivaScience, Oakville, ON, Canada).
Mice and Islet Isolation
C57Bl/6 (Bl/6), Balb/c, and FVB mice were purchased from Jackson Labs (Bar Harbor, ME, USA). Animals were housed in the Child and Family Research Institute animal facility and cared for in accordance with the guidelines of the Canadian Council on Animal Care and regulations of the University of British Columbia. Pancreatic islets were isolated from 12-week-old donor mice (Bl/6, FVB, or Balb/c, males and females) by ductal collagenase (Sigma-Aldrich, Oakville, ON, Canada) injection, digestion, and purification with filtration method as described previously (23,26). Bl/6 islets used for primary transplant were incubated overnight at 37°C in RPMI-1640 (Gibco, Burlington, ON, Canada) supplemented with 10% FBS (Gibco), 100 units/ml penicillin (Sigma-Aldrich), 1% Glutamax (Gibco) with either Ad-CCL22 or Ad-LacZ, using a multiplicity of infection (MOI) of 10.
Primary and Secondary Islet Transplantation
Five days before transplantation, female recipient mice were rendered hyperglycemic by a single intraperitoneal injection of 250 mg/kg (Balb/c) or 180 mg/kg (Bl/6) of streptozotocin (STZ) (Sigma-Aldrich) in acetate buffer pH 4.5 [0.08 M glacial acetic acid (Fisher, Ottawa, ON, Canada), 0.04 M sodium acetate (Fisher), and 0.15 M sodium chloride (Fisher)]. Nonfasting tail vein blood glucose was measured twice weekly using OneTouch Ultra 2 (LifeSpan, Milpitas, CA, USA). Mice were considered diabetic if blood sugar was >22 mM for two consecutive measurements before transplantation. Islets were transplanted into the left renal subcapsular space of recipient mice under isofluorane (Abbott Laboratories, Saint-Laurent, QC, Canada) anesthesia as previously described (11,23). The time of graft failure was determined by two consecutive blood glucose measurements >15 mM, at which point diabetic mice were sacrificed. Removal of the graft-bearing kidney was performed on recipients of Ad-CCL22-transduced islets to ensure that normoglycemia was graft dependent. To assess immunological tolerance, mice with blood glucose levels >20 mM after nephrectomy between days 70 and 80 were retransplanted the following day with the same donor strain (Bl/6) or third-party (FVB) islets into the right renal subcapsular space.
Graft Function
In vivo graft function was assessed by intraperitoneal glucose tolerance test. Mice were fasted for 5 h and injected with 2 g/kg body weight D50 glucose (Baxter Healthcare, Mississauga, ON, Canada). Tail blood glucose was measured at 0, 15, 30, 60, 90, and 120 min. Naive, age-matched Balb/c mice were used as controls.
CCL22 Protein Determination
CCL22 was determined in transduced islets by Western blot. Briefly, 10 μg protein from lysed islets was boiled with a reducing sample buffer [0.1 M Tris-HCl (Fisher), 4% SDS (Bio-Rad, Hercules, CA, USA), 0.15% bromo-phenol blue (Fisher), 24% glycerol (Sigma-Aldrich), 5% β-mercaptoethanol (Sigma-Aldrich)] and resolved on a 20% SDS-polyacrylamide gel (Bio-Rad) at 100 volts. The gel was transferred onto Immobilon-P membranes (Millipore, Billerica, MA, USA) at 100 volts for 1 h using the Trans- Blot System (Bio-Rad). Immunoblotting was performed using polyclonal anti-CCL22 antibody (1:100; R&D Systems, Minneapolis, MN, USA) and detected by chemiluminescence (Amersham, Quebec, QC, Canada). CCL22 protein levels in serum, media, and grafts were measured by ELISA (R&D Systems) following the manufacturer's instructions.
Real-Time PCR
To quantify viral gene expression of CCL22, total RNA was extracted from islet grafts using RNeasy mini kit (Qiagen, Germantown, MD, USA) in accordance with the manufacturer's instructions. cDNA synthesis to analyze viral expression of CCL22 was performed using SuperScript III Reverse Transcriptase (Invitrogen, Burlington, ON, Canada) with 10 μM Oligo-dT primer (Invitrogen). PCR reactions were performed in duplicate using the Applied Biosystems 7500 System with EvaGreen Mastermix (Applied Biosystems, Burlington, ON, Canada). Primers used were as follows: CCL22 (111 bp) Fw 5′-AGGTCCCTATGGTGCCAATGT-3′ and Rv 5′-CGGCAGGATTTTGAGGTCCA-3′; GAPDH (123 bp) Fw 5′-AGGTCGGTGTGAACGGATTTG-3′ and Rv 5′-TGTAGACCATGTAGTTGAGGTCA-3′. Relative quantification was performed using GAPDH as a housekeeping gene. Gene expression was calculated using the DDCT method.
Histological Analysis of the Pancreas
Pancreata and grafts were harvested and fixed in 4% paraformaldehyde (Sigma-Aldrich) overnight. Tissues were embedded in paraffin, and 5-μm sections were immunostained using guinea pig anti-insulin and rabbit anti-glucagon (1:100; Dako, Burlington, ON, Canada) as primary antibodies, and goat anti-guinea pig Alexa 594 and goat anti-rabbit Alexa 488 (1:100; Molecular Probes, Indianapolis, IN, USA) as secondary antibodies. Leukocyte immunostaining was performed following antigen retrieval by steaming 30 min in Dako Target Retrieval Solution (Dako). Sections were stained using rat anti-mouse CD45 (1:25; BD Pharmingen, Mississauga, ON, Canada) or anti-mouse FoxP3 (1:100; eBioscience, San Diego, CA, USA) and anti-rat Alexa 488 (1:100, Molecular Probes) as secondary antibody. Images were captured using an Olympus Bx61 microscope and In Vivo or DP controller software.
Flow Cytometry
Islet grafts, lymph nodes, and spleens were harvested and strained through a 40-μm filter (BD Biosciences, Mississauga, ON, Canada) and washed before incubation with antibodies. Spleens were treated for 5 min with red blood cell (RBC) lysis buffer containing 155 mM NH4Cl (Sigma-Aldrich), 12 mM NaHCO3 (Fisher), and 0.1 mM EDTA (Fisher). Monoclonal antibodies to CD8 (1:200; 53-6.7), CD4 (1:200; RM4-5), CD3 (1:250; 145-2C11), IgG1 (1:100), and IgG2 (1:100) were purchased from BD Biosciences. Antibodies to FoxP3 (1:100; clones MF23, MF-14, and 150D) and CCR4 (1:100; 2G12) were obtained from BioLegend (San Diego, CA, USA). Monoclonal antibody to Ki-67 (1:100; SolA15) was purchased from eBioscience. For intracellular staining, cells were fixed and permeabilized using the Cytofix/ Cytoperm kit (BD Biosciences). Countbright Absolute Counting Beads (Invitrogen) were used to calculate the absolute number of cells within the graft. For alloantibody measurement, sera from Balb/c recipients of CCL22- or LacZ-expressing islets were collected at 20 and 60 days posttransplantation. Serum was incubated with allogeneic (Bl/6) splenocytes for 1 h. Splenocytes were washed, incubated with anti-IgG1 or anti-IgG2 antibodies for 30 min, and specific antidonor antibodies bound to splenocytes were measured by flow cytometry. Background IgG levels were assessed by incubating serum with syngeneic (Balb/c) splenocytes. A FACSCalibur cytometer (BD Biosciences) and CellQuest software were used for data acquisition. Data were analyzed using FlowJo7.2.5 software. Mean fluorescence intensity of total antidonor IgG was normalized to background levels in serum incubated with Balb/c splenocytes.
Anti-CD25 Antibody Treatment
Diabetic Balb/c mice transplanted with islets expressing CCL22 were injected intraperitoneally twice weekly with 100 μg anti-CD25 monoclonal antibody (PC61) (AbLab, Vancouver BC, Canada) or rat IgG isotype control (AbLab) starting at day of transplantation. Treg depletion was assessed weekly by FACS analysis of CD4-, CD25-, and FoxP3-expressing cells in peripheral blood.
Mixed Leukocyte Assay
Splenocytes were isolated from recipients of CCL22-transduced islets at 60 days posttransplantation and compared with splenocytes from nontransplanted Balb/c control mice. Responder cells were seeded at 2 × 105 cells/well in triplicate and stimulated with varying numbers of irradiated (30 Gy) Bl/6 splenocytes in a final volume of 200 μl RPMI (Gibco) supplemented with 10% FBS (Gibco). After 4 days of culture, 1 μCi/well of [3H]methyl thymidine (Amersham) was added for the last 16 h. Cells were collected using a cell harvester (Tomtec, Hamden, CT, USA) and thymidine incorporation measured in a Microbeta Trilux scintillation counter (PerkinElmer, Waltham, MA, USA).
Statistical Analysis
Graft survival curves were generated using Kaplan– Meier life-table analysis (Prism, La Jolla, CA, USA), and the survival curves were compared using log-rank test. Statistical analysis was performed using Student's two-tailed t-test with Welch's correction for unequal variance. Differences were considered significant if p < 0.05. Data in bar graphs are represented as mean ± SEM.
Results
Islet Expression of CCL22 Protects From Islet Allograft Rejection
CCL22 expression in Bl/6 mouse islets following overnight transduction with Ad-CCL22 was confirmed by ELISA of islet incubation media, showing increased CCL22 secretion with increasing MOI of Ad-CCL22. MOI of 10 was chosen for transplant experiments, based on maximal CCL22 production in the absence of detectable islet cell toxicity or dysfunction as assessed by glucose-stimulated insulin secretion (data not shown). To determine the effect of CCL22 expression on islet allograft survival, we induced experimental diabetes in Balb/c mice using STZ. Bl/6 mouse islets were isolated and transduced overnight with Ad-CCL22, Ad-LacZ, or left untreated. Islets (300 per recipient) were transplanted under the renal capsule of age-matched, STZ-diabetic MHC-mismatched Balb/c recipients. Blood glucose levels were measured in transplant recipients weekly to determine return of diabetes. At 80 days posttransplant, 75% of mice transplanted with CCL22-expressing islets remained normoglycemic, whereas all Ad-LacZ-transduced and nontransduced grafts were rejected by 60 days (Fig. 1A). Recipients of CCL22-expressing islet allografts maintained normal glucose tolerance (Fig. 1B) 60 days posttransplantation.

CCL22 expression protects islet allografts. (A) STZ-treated Balb/c mice (H2d) were transplanted with 300 Bl/6 (H2b) islets previously transduced with Ad-CCL22 (solid line, n = 11), Ad-LacZ (dashed line, n = 9), or not transduced (dotted line, n = 8). Left panel shows blood glucose measurements with nephrectomy performed after day 80 in surviving mice. Kaplan–Meier survival curve (right panel) showed prolonged survival of islet allografts expressing CCL22 (p < 0.0001 vs. LacZ control). (B) Intraperitoneal glucose tolerance test in STZ-diabetic Balb/c mice transplanted with Ad-CCL22 islets (n = 8, solid line) 60 days posttransplantation or in non-diabetic, nontransplanted age-matched control mice (n = 5, dashed line) (p = NS). Data are means ± SEM.
Preservation of β-Cells in CCL22-Expressing Grafts
At 20 days posttransplant, CCL22 protein was detected in islet grafts as determined by Western blot (Fig. 2A) and quantified by ELISA (Fig. 2B). CCL22 levels in serum were not significantly elevated compared to LacZ controls, emphasizing a local gradient effect of CCL22 in transplanted islets (data not shown). CCL22 mRNA was also detectable at 15 days posttransplant in islet grafts transduced with Ad-CCL22, but was decreased to 10% of these levels at 60 days posttransplant, probably due to loss of adenoviral expression in the graft (21) (Fig. 2C). Immunohistochemical analysis of islet allografts 15 and 60 days after transplantation revealed that, whereas CCL22-expressing islet grafts maintained normal islet structure with numerous insulin-immunopositive cells, control grafts showed almost complete absence of insulin production by β-cells (Fig. 3). Both CCL22- and LacZ-expressing grafts were surrounded by an intense CD45+ leukocytic infiltration, but insulin-positive area was detectable and still clearly preserved at 60 days following transplantation of CCL22-expressing islets (Fig. 3).

CCL22 expression in isolated islet grafts. Determination of CCL22 protein expression 20 days after transplantation in excised grafts by (A) Western blot or (B) ELISA (n = 4). (C) CCL22 mRNA levels in grafts harvested at 15 or 60 days following transplantation (n = 3, black bars) were analyzed by qPCR and normalized to LacZ-transduced controls (n = 3, white bars). ***p < 0.0001 versus LacZ control. *p < 0.05 versus LacZ controls. Data are means ± SEM.

CCL22 preserves insulin-positive cells in islet grafts. Sections of islet allografts at 15 and 60 days posttransplant were assessed histologically by immunostaining for insulin (red) and leukocyte infiltration (CD45; green). Note the presence of intact islets containing numerous insulin-immunopositive cells in Ad-CCL22-expressing grafts but not in LacZ-expressing control grafts. Scale bar: 100 μm.
Recruitment of Tregs in CCL22-Expressing Islet Allografts
To determine whether CCL22 attracted Tregs, islet graft sections were immunostained for the presence of the Treg marker FoxP3, 15 days after transplantation. CCL22-expressing islet allografts had a higher frequency of Tregs compared to islets transduced with Ad-LacZ (Fig. 4A). To confirm this observation, islet grafts were dispersed to single cells and analyzed by flow cytometry. A larger proportion of CD4+FoxP3-expressing cells (Tregs) was observed in CCL22-expressing grafts at 15 days compared to Ad-LacZ transduced grafts (Fig.4B) as well as an increase in Treg cell numbers (Fig. 4C). CD4+FoxP3+ cells did not exhibit an increase in Ki-67 expression, indicating there is no increased proliferation of Tregs within CCL22-transduced islets. There was no difference in the proportion of CCR4+ Tregs in CCL22 grafts compared to LacZ grafts, nor was there an increase in cell surface expression of CCR4 on recruited Tregs (data not shown). Proportions and absolute numbers of CD4+ and CD8+ were not significantly different at day 15 in CCL22-expressing mice when compared to LacZ controls (Fig. 4E, F), suggesting that Tregs are specifically and preferentially recruited to CCL22-expressing islets. The proportions of CD4+FoxP3+ T cells and of CD4+ and CD8+ cells decreased after day 15 posttransplant, likely due to loss of adenoviral CCL22 expression (20) (Figs. 2B, C and 4B, E, F). Recruitment of the Treg population appeared to be specific to the islet graft, since there was no observable change in the proportion of CD4+ T cells or Tregs in the draining (renal) lymph nodes or spleens of recipients of CCL22-expressing islet grafts (data not shown).

Adenoviral expression of CCL22 attracts CD4+FoxP3+ Tregs to islet allografts.(A) Immunostaining of graft sections at 15 days posttransplant showing increased frequency of FoxP3+ cells (brown, black arrows) in CCL22-expressing grafts compared to LacZ control. Scale bar: 100 μm. (B-D) Flow cytometric analysis of dispersed single cells from islet grafts harvested at 15 (B-D) or 50 days (B) posttransplant. (B, C) The percentage and absolute number of CD4+ cells that are FoxP3+ in islet grafts was significantly elevated at 15 days posttransplantation in CCL22-expressing grafts (black bar; n = 3) compared to LacZ-expressing grafts (white bar; n = 3). (D) There was no difference in expression of Ki-67 between CD4+FoxP3+ Tregs in CCL22-expressing grafts and those from LacZ-expressing grafts at 15 days posttransplantation. (E, F) Percentage and absolute number of CD4+ and CD8+ cells was not significantly different at 15 days after transplantation. *p<0.05 versus LacZ control. All experiments were conducted in triplicate.
To determine whether the protective effect of CCL22 expression was due to recruited Tregs, anti-CD25 antibody (Ab) was administered to recipients of CCL22-expressing islet allografts starting on the day of transplantation. Anti-CD25 Ab efficiently depleted Tregs in vivo (Fig. 5A). In all recipients (five of five) treated with anti-CD25 Ab, CCL22-expressing allografts were rejected (as determined by return of hyperglycemia) 45 days posttransplantation, whereas all recipients (four of four) of CCL22-expressing islets treated with rat IgG isotype control remained normoglycemic for more than 60 days (Fig. 5B). These data indicate that recruitment of Tregs plays an essential role in CCL22 protection of islet allografts.

Depletion of CD25-expressing cells abrogates CCL22′s protective action on islet allografts. STZ-diabetic Balb/c mice bearing C57Bl/6 CCL22-expressing grafts were given weekly injections of either anti-CD25 Ab (clone PC61) or isotype rat IgG control. (A) Rapid and sustained depletion of blood Tregs following anti-CD25 Ab treatment (solid line, n = 5) relative to those treated with IgG isotype control (dashed line, n = 4). The proportion of peripheral blood CD4+ T cells that express FoxP3+ was quantitated by flow cytometry in Ab-treated recipient mice of CCL22-expressing islet allografts. (B) Kaplan–Meier survival curve of glucose levels showing that CCL22-expressing grafts are rejected in recipients of anti-CD25 Ab (solid line, n = 5) but not in recipients of control IgG Ab (dashed line, n = 4). p < 0.0001 versus IgG isotype Ab control.
CCL22 Induces Donor-Specific Tolerance to Fully Mismatched Islet Allografts
We next analyzed allospecific reactivity in graft recipients by measuring antidonor antibodies, comparing levels of IgG from serum of graft recipients that bound donor strain (Bl/6) splenocytes to levels bound to syngeneic (Balb/c) splenocytes. Whereas Balb/c recipients of Ad-LacZ-transduced Bl/6 islets showed robust alloantibody production, recipients of CCL22-expressing Bl/6 islets produced little to no detectable alloantibodies at 20 and 60 days posttransplantation (Fig. 6A). In addition, splenocytes from recipients of CCL22-expressing islet grafts showed no proliferative response in mixed lymphocyte reactions to donor splenocytes at 60 days posttransplantation (Fig. 6B). The indefinite protection seen in most CCL22-expressing islet allografts (despite diminishing CCL22 expression), together with the lack of alloantibodies and splenocyte proliferation in mixed lymphocyte reactions, suggests that continuous CCL22 expression and Treg recruitment is not required for islet allograft protection, but rather that tolerance may be induced in this model.

CCL22 diminishes alloantigen-specific antibody response and allogeneic splenocyte proliferation. (A) Specific antidonor antibodies were measured in serum of CCL22-expressing recipients (black line) and Lac-Z-transduced controls (gray line with arrows) after binding to Bl/6 splenocytes. Alloantibody measurements were normalized to background levels in serum incubated with syngeneic Balb/c splenocytes (gray shadow and red dotted line) at day 20 (n = 4 each group) and at day 60 (CCL22 n = 4, LacZ n = 2). Representative histogram plots (left panel) and cumulative measurements (right panel) are shown. *p < 0.05 versus LacZ control, p = NS versus allogeneic control. Experiments were conducted in duplicate. (B) Allostimulated in vitro proliferation of splenocytes isolated from recipients of CCL22-expressing islet grafts (n = 3; solid line) and naive controls (n = 3; dashed line). Splenocytes from CCL22 recipients were stimulated with indicated ratios of irradiated Bl/6 splenocytes and pulsed with [3H]thymidine for a 16-h period at the end of 4 days of culture. Results are expressed as counts per minute (cpm; mean ± SEM). **p < 0.001, *p < 0.05 versus naive splenocytes. Experiments were conducted in triplicate.
To assess donor specificity of tolerance, we transplanted STZ-diabetic Balb/c recipients with Ad-CCL22-transduced Bl/6 islets, and following 70 to 80 days of normoglycemia, removed the primary graft and transplanted 300 untreated Bl/6 (n = 4) or third-party (n = 3; completely H2-disparate from both Balb/c and Bl/6) islets beneath the contralateral kidney capsule. Mice transplanted with untreated Bl/6 islets maintained normoglycemia for more than 230 days (Fig. 7A), whereas recipients of third-party (FVB) islets rejected the graft in less than 3 weeks (Fig. 7A). In addition, immunohistochemical analysis of the grafts revealed that the second islet transplant in previous recipients of CCL22-expressing grafts maintained normal islet structure and lack of immune infiltration (Fig. 7B). These findings indicate that alloantigen-specific tolerance can be elicited through the action of CCL22.
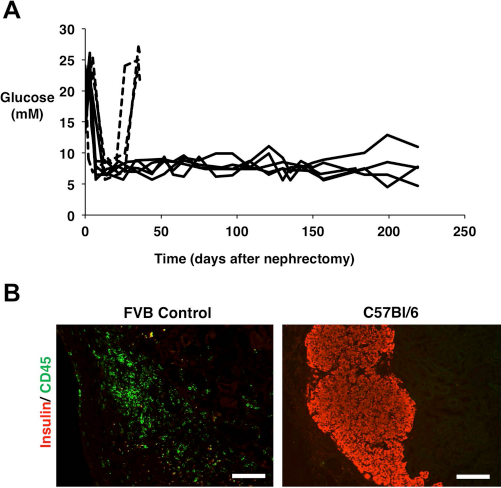
CCL22 induces unresponsiveness to second donor islet allografts but not to third-party islet transplants. (A) Blood glucose levels in Balb/c recipients of CCL22-expressing Bl/6 islet allografts that received a second islet allograft between 70 and 80 days following the first transplant. After removal of the primary graft, mice with blood glucose >20 mM for two consecutive measurements were retransplanted at time zero with untreated Bl/6 islets (n = 5, solid line) or third-party FVB (H2q) islets (n = 3, dashed line). Recipients of CCL22-expressing grafts become tolerant to Bl/6, but not to FVB antigens. (B) Preservation of insulin-positive cells and absence of immune infiltration in a representative Bl/6 second transplant graft at 225 days posttransplant, but not in an FVB islet second transplant graft at the time of rejection. Graft sections were immunostained for insulin (red) or CD45 (green). Scale bar: 100 μm.
CCL22 Expression Protects From Rejection in a More Stringent Islet Allograft Model
The effect of CCL22 on preventing a more aggressive islet alloimmune response was investigated using Bl/6 mice as recipients and Balb/c mice as islet donors (16). Balb/c islets were isolated and transduced overnight with Ad-CCL22 or Ad-LacZ. As before, 300 islets were transplanted under the renal capsule of age-matched, STZ-diabetic MHC-mismatched Bl/6 recipients. Similar to the results obtained in the reverse setting, more than 60% of mice transplanted with CCL22-expressing islets remained normoglycemic at day 50 posttransplantation, well after loss of CCL22 expression in the graft. By 80 days, only 30% of mice maintained normal glucose levels. In contrast, however, all Ad-LacZ-transduced grafts were rejected by 30 days (Fig. 8A). In addition, immunohistochemical analysis of the grafts at 60 days posttransplant revealed that CCL22-expressing grafts in Bl/6 recipients maintained normal islet structure and showed lack of immune infiltration when compared to a rejecting LacZ control at 30 days (Fig. 8B).

CCL22 expression protects from allograft rejection in a more stringent model. (A) STZ-induced Bl/6 mice were transplanted with 300 Balb/c islets previously transduced with Ad-CCL22 (solid line, n = 6) or Ad-LacZ (dashed line, n = 3). Kaplan–Meier survival curve showed prolonged survival of islet allografts expressing CCL22 (p < 0.0001 vs. LacZ control). (B) Immunostaining of graft sections stained for insulin (red) and CD45 (green) in Bl/6 recipients transplanted with Balb/c islets expressing CCL22 (at day 60) or LacZ (at time of rejection). Scale bar: 100 μm.
Discussion
Expression of CCL22 in islet allografts leads to Treg recruitment and long-term protection from rejection in MHC-mismatched, diabetic recipients. These findings extend our recent findings that islet CCL22 expression delays both spontaneous and recurrent autoimmune diabetes in NOD mice (21), by showing that CCL22 also protects islet allografts.
An increased proportion of FoxP3+ cells was observed in grafts expressing CCL22, demonstrating that endogenous Tregs are recruited specifically to islet allografts by CCL22 expression. Interestingly, the proportion of CD4+ cells was slightly increased in the graft, likely due to CCR4 expression on activated CD4+ cells (20). This finding is consistent with a recent description of Tregs trafficking to the same sites as cells they suppress, in models not dependent on exogenous chemokine expression (18). A previous report suggests that transgenic lifetime expression of CCL22 in islets in NOD mice decreases the time to diabetes onset (13); in our studies, in contrast, we have induced CCL22 expression in transplanted islets in adult animals. Our findings suggest that the time and conditions of expression of this chemokine may greatly influence the outcome of disease. Recruitment of Tregs to the graft by CCL22 at an early stage of transplantation is likely important for not only induction but also long-term maintenance of tolerance to allogeneic tissue.
The changes in immune cell populations induced by islet expression of CCL22 were localized to the islet graft, with no changes found in the blood, spleen, or draining lymph nodes. This is not unexpected, since the graft populations are extremely small when compared to the total body numbers of these cells. Depletion of Tregs in recipients of CCL22-expressing islet allografts, by administration of anti-CD25 antibody, induced graft rejection, supporting the idea that the protective effect of CCL22 was mediated by recruitment of CD25-expressing cells, presumably Tregs. We similarly observed a requirement for CD25-expressing cells in CCL22-mediated protection of syngeneic islet transplants from recurrent autoimmunity in diabetic NOD mice (21).
Our data suggest that continued CCL22 expression is not necessary for prolonged protection from islet allograft rejection, since allograft rejection does not occur at a time when adenoviral CCL22 expression has decreased to undetectable levels (21). In addition, since the alloantibody response and donor-specific mixed lymphocyte response in recipients of CCL22-expressing grafts are abrogated, continued expression of CCL22 is likely not essential for continued graft protection. These observations, and the prolonged protection seen in most CCL22-expressing islet allografts (despite diminishing expression of CCL22), suggest that tolerance may be induced in this model. In support of this idea, when untreated islets of same donor origin were transplanted into the contralateral kidney of previous recipients of CCL22-expressing islet allografts, the second islet transplants survived and maintained normoglycemia in recipients for more than 230 days. Importantly, when MHC-disparate third-party islets were transplanted into such recipients, the mice rapidly rejected the grafts, demonstrating donor specificity of immune tolerance. The observed rejection of third-party islet grafts further suggests that induction of tolerance by CCL22 is not a consequence of a general loss of allore-sponsiveness, but that recruited Tregs may participate in tolerance induction by suppression of allo-specific T cells or other alterations in graft microenviroment. These findings point to the potential therapeutic value of CCL22 and support previous findings that suggest that suppressive Tregs play an important role in tolerance induction (4,10,15,17)
Balb/c mice are well known to develop T-helper cell type 2 (Th2)-dominated responses, in contrast to Bl/6 mice, which are more prone to Th1 immune responses that result in more aggressive alloimmune rejection (27). Even in the more stringent model, transplanting Balb/c islets into Bl/6 recipients, CCL22 conferred protection from allograft rejection for over 60% of mice until day 60 posttransplantation, although the protection observed was not as marked as in the less stringent setting using Balb/c recipients, with only 30% of mice maintaining normoglycemia past 80 days. Since CCL22 protects from recurrent autoimmune destruction of syngeneic islet transplants in diabetic NOD mice (21), it will now be of importance to show whether this approach can also be used to protect islet allografts in diabetic autoimmune recipients.
A major goal in islet transplantation is induction of tolerance; alloreactivity is controlled, grafts function normally, and the recipient is liberated from the side effects of global immunosuppression. The importance of Tregs in generating and maintaining tolerance has been described in numerous other studies that show an association of Tregs with long-term graft acceptance (2,3,14,22). Although phase I clinical studies involving Treg cell therapy are already under way (24), there are still hurdles to be overcome prior to application of this strategy to the treatment of human type 1 diabetes and in islet transplantation. Tregs represent less than 1% of peripheral blood mononuclear cells and, for current therapeutic studies, need to be expanded in vitro prior to clinical application (2,24). Since conventional CD4 cells grow much more readily than Tregs, and obtaining pure populations of human Tregs is problematic, this in vitro procedure carries the risk of resultant infusion of conventional, potentially pathogenic T cells. A distinct advantage of our approach is that, by stimulating recruitment of endogenous Tregs directly to the graft (6), it potentially avoids this risk and additionally directs the attention of the regulatory cells to the target of immune attack, rather than risking the potentially less focused result of Treg infusion. Even if it were to prove ineffective as monotherapy in patients, localized CCL22 overexpression or delivery might prove a significant potentiator of Treg therapy, reducing the amount of Treg expansion required prior to cell infusion.
In summary, we have observed that expression of CCL22 in islet cells by ex vivo adenoviral transduction of isolated islets confers protection of these cells from allograft rejection, without impairing β-cell function. Our findings point to the tremendous therapeutic potential of CCL22 and the urgent need to understand more precisely how CCL22, and possibly other chemokines, may protect β-cells from immune attack. Manipulating the migration of endogenous Tregs by viral or nonviral gene therapy approaches may be a novel therapeutic strategy not only for diabetes but also for other autoimmune diseases and organ transplantation.
Footnotes
Acknowledgments
This work was supported by a grant from the JDRF (1-2011-550) to C.B.V. and from the Canadian Institutes of Health Research (CIHR MOP-115199) to M.K.L. Core support was provided by a donation from the Canucks for Kids Fund (CFKF) to the CFKF Childhood Diabetes Laboratories. M.K.L. holds a Canada Research Chair in Transplantation. J.M., L.B., M.O., and S.A. are supported by trainee awards from JDRF (J.M., L.B.), the CIHR Transplant Training Program (J.M., L.B., M.O., S.A.), and the Child and Family Research Institute (M.O.). G.H. is supported by a fellowship from Michael Smith Foundation for Health Research (MSFHR). We wish to acknowledge Dr. Tim Kieffer (University of British Columbia) for providing Ad-LacZ vector. We thank members of the Verchere laboratory for the helpful discussion and M. Komba for technical assistance. J.M., M.O., S.A., L.B., D.D., G.S., and G.H. performed experiments and acquired, analyzed, and interpreted data. J.P., R.T., M.K.L., and P.O. facilitated experiments through the provision of reagents and interpreted the data. R.T. and C.B.V. designed the study and supervised the project. J.M. and C.B.V. wrote the manuscript. All authors contributed to discussions and critically edited, reviewed, and gave final approval of this version of the manuscript to be published. The authors declare no conflicts of interest.