
Abstract
Bone marrow-derived mesenchymal stem cells (MSCs), which have beneficial effects in acute lung injury (ALI), can serve as a vehicle for gene therapy. Angiotensin-converting enzyme 2 (ACE2), a counterregulatory enzyme of ACE that degrades angiotensin (Ang) II into Ang 1–7, has a protective role against ALI. Because ACE2 expression is severely reduced in the injured lung, a therapy targeted to improve ACE2 expression in lung might attenuate ALI. We hypothesized that MSCs overexpressing ACE2 would have further benefits in lipopolysaccharide (LPS)-induced ALI mice, when compared with MSCs alone. MSCs were transduced with ACE2 gene (MSC-ACE2) by a lentiviral vector and then infused into wild-type (WT) and ACE2 knockout (ACE2-/y) mice following an LPS-induced intratracheal lung injury. The results demonstrated that the lung injury of ALI mice was alleviated at 24 and 72 h after MSC-ACE2 transplantation. MSC-ACE2 improved the lung histopathology and had additional anti-inflammatory effects when compared with MSCs alone in both WT and ACE2-/y ALI mice. MSC-ACE2 administration also reduced pulmonary vascular permeability, improved endothelial barrier integrity, and normalized lung eNOS expression relative to the MSC group. The beneficial effects of MSC-ACE2 could be attributed to its recruitment into the injured lung and enhanced local expression of ACE2 protein without changing the serum ACE2 levels after MSC-ACE2 transplantation. The biological activity of the increased ACE2 protein decreased the Ang II amount and increased the Ang 1–7 level in the lung when compared with the ALI and MSC-only groups, thereby inhibiting the detrimental effects of accumulating Ang II. Therefore, compared to MSCs alone, the administration of MSCs overexpressing ACE2 resulted in a further improvement in the inflammatory response and pulmonary endothelial function of LPS-induced ALI mice. These additional benefits could be due to the degradation of Ang II that accompanies the targeted overexpression of ACE2 in the lung.
Keywords
Introduction
Acute lung injury (ALI) is a devastating clinical syndrome that is characterized by the diffuse damage of lung vascular endothelial cells and alveolar epithelial cells and an excessive inflammatory response in the lung (2,5,40). There are currently few specific pharmacological therapies that attenuate ALI and promote lung repair (6,10,18,35).
Experimental and clinical evidence indicates that the renin–angiotensin system (RAS) plays an essential role in the pathogenesis of ALI through an increased level of angiotensin (Ang) II generated by the angiotensinconverting enzyme (ACE). The main active molecule, Ang II, can initiate lung inflammatory responses and impair the function of the pulmonary endothelial barrier via the Ang II type 1 receptor (AT1R) (16,17,23,27,45). ACE2, the first homolog of ACE (a counterregulatory αenzyme of ACE), degrades Ang II into Ang 1–7 and mitigates the detrimental effects of Ang II in ALI animal models (16,17). Moreover, previous studies have found that ACE2 mRNA, protein, and enzymatic activity were severely downregulated in human and experimental lung tissue injuries (17,24). In ACE2 knockout mice, the loss of ACE2 results in higher Ang II levels and a more severe lung injury, indicating that the decrease of ACE2 is a major factor contributing to the pathogenesis of ALI by permitting Ang II accumulation. Accordingly, therapies aimed at increasing ACE2 expression in lung tissue might attenuate ALI. However, the systemic infusion of recombinant ACE2 decreases serum Ang II and may affect blood pressure (37,44). Therefore, we hypothesized that a treatment with ACE2 targeted specifically to the injured lung would achieve the optimal therapeutic effect.
In animal models of lung injury, bone marrow-derived mesenchymal stem cells (MSCs) have been shown to attenuate the severity of lung injury and to reduce mortality. These effects occurred through several mechanisms, including differentiation into mature lung epithelial and endothelial cells and a paracrine function to modulate localized inflammation (11,21,33,42). Importantly, MSCs can also act as a vehicle for delivering a protective gene by overexpressing a transgene at the injured site, which could not only enhance its therapeutic effects but also promote local lung repair (29). Therefore, the combination of MSCs and the protective gene ACE2 may be a potential strategy for the treatment of ALI.
In the present study, we tested the hypothesis that MSCs with the stable long-term expression of ACE2 via lentiviral-mediated gene transfer could be targeted to the injured lung, locally overexpress ACE2, and attenuate lung injury by reducing the inflammatory response and improving the function of the lung vascular endothelium in lipopolysaccharide (LPS)-induced ALI mice.
Materials and Methods
Ethics Statement
Male wild-type (WT) C57BL/6 mice (Laboratory Animal Center, Academy of Military Medical Sciences, Beijing, China) and male ACE2 knockout C57BL/6 (ACE2-/y) mice (Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences, Beijing, China) were maintained under specific pathogen-free conditions. All experiments involving the use of animals were approved by the Committee of Animal Care and Use of Southeast University.
Production of Lentiviral Vectors and Transduction of MSCs
MSCs from the bone marrow of male WT C57BL/6 mice and 293FT cells were purchased from Cyagen Biosciences, Inc. (Guangzhou, China) as previously described (25). MSCs from passages 4–7 were used for transduction. The Gateway cloning system, as previously reported (12), was used to transfer the full-length coding sequence of ACE2 (NM_001130513.1, 2418 bp) into the human translation elongation factor 1a (EF1a) promoter-dependent lentiviral expression vector pLV.EX3d.P/neo (Cyagen Biosciences, Inc., Guangzhou, China). The ACE2 gene was cloned into the lentiviral expression vector between the EF1a promoter and the internal ribosomal entry site (IRES)-dependent eGFP via the BP reaction (Gateway® BP Clonase” II Enzyme Mix; Invitrogen Life Technologies, Carlsbad, CA, USA) and the LR reaction (Gateway® LR Clonase” II Plus Enzyme Mix; Invitrogen Life Technologies). The lentivirus was packaged in 293FT cells with the aid of three packaging plasmids: pLV/helper-SL3, pLV/helper-SL4, and pLV/helper-SL5 (http://www.addgene.org). Subsequently, a high titer of the recombinant lentivirus was obtained and used to transfect the MSCs. MSCs carrying either eGFP (MSC-GFP) alone or both ACE2 and eGFP (MSC-ACE2) were harvested after selection using G418 (0.5 mg/ml; Amresco, Solon, OH, USA) for 7–14 days. The transduction efficiency was evaluated by detecting the expression of eGFP with an Olympus IX51 fluorescence microscope (Olympus Co., Tokyo, Japan) and a Becton Dickinson FACS Calibur flow cytometer (FACS Calibur; Becton-Dickinson, Mountain View, CA, USA). The transcription of the ACE2 transgene was evaluated using reverse transcription-polymerase chain reaction (RT-PCR). The expression of ACE2 protein was determined using Western blot analysis, and the secreted ACE2 protein in the culture media was evaluated using an enzyme-linked immunosorbent assay (ELISA) kit (Shanghai Westang Bio-tech. Co., Ltd., Shanghai, China). The primers used for designing the RT-PCR were based on the sequences of the genomic clones: ACE2 (254 bp): forward, 5′-TGGTAGTGGTTGGCATCATCATCC-3′ and reverse, 5′-ACGCACACCGGCCTTATTCC-3′ β-actin (243 bp): forward, 5′-ATCGTGGGCCGCCCTAGGCA-3′ and reverse, 5′- TGGCCTTAGGGTTCAGGGGGG-3′. The primary antibodies against ACE2 (1:100 dilution; Abcam Ltd., Cambridge, UK) and β-actin (1:3,000 dilution; Bioworld Technology, Co. Ltd., Nanjing, China) were used in the Western blot analysis. The cells in passages 7–10 were used for the in vivo experiments.
Murine Model of LPS-Induced ALI
Eight- to 10-week-old WT and ACE2-/y mice received intratracheal instillations of 100 μg of LPS (Escherichia coli 0111:B4; Sigma-Aldrich, St. Louis, MO, USA) dissolved in 50 μl of phosphate-buffered saline (PBS; Wisent Inc., St-Bruno, Quebec, Canada) as previously described to produce the ALI model (9). PBS, MSC-GFP, and MSC-ACE2 (5 × 105 cells resuspended in 100 μl PBS) (42) were infused via tail vein injection 4 h (11) after the LPS challenge. Mice without the LPS instillation were injected with PBS as a control. Either 24 or 72 h after the MSC treatment, the mice were sacrificed, and tissues were collected for further analysis.
Tracking of MSCs in the Lung
MSC-ACE2 cells (5 × 105) labeled with CellVue NIR815 dye (eBioscience Inc., San Diego, CA, USA) were transplanted into WT ALI mice. Three mice at each time point (30 min, 24 h, and 72 h postinjection) were imaged for whole-body and ex vivo organs, including the lung, heart, kidney, spleen, pancreas, small intestine, and liver using a Maestro In-Vivo Optical Imaging System (excitation = 786 nm; emission = 814 nm; exposition time = 4,000 ms; Caliper Life Sciences, Woburn, MA, USA) (28,38). The autofluorescence spectra were then unmixed based on their respective spectral patterns using Maestro 2.4 software (Caliper Life Sciences). The fluorescence intensity of each organ was measured by placing the ROIs on the organ of interest, and the average signals were normalized to the exposure time and the area of the ROI (scaled counts/s) (28,38).
Fluorescence microscopy of the lung sections from the control, ALI, MSC-GFP, and MSC-ACE2 groups of WT mice (n = 3 per group) at 72 h was also performed to detect the expression of eGFP and confirm the localization of the MSC-ACE2 cells. Briefly, the right lower lobes of the mice were collected and fixed in 4% paraformaldehyde overnight at 4°C. The tissues were then frozen in optimal cutting temperature medium (Sakura Finetek USA, Inc., Torrance, CA, USA) and cut into 5-μm-thick sections. The nuclei were stained with 4,6-diamidino-2-phenylindole (Sigma-Aldrich), and eGFP fluorescence was monitored using an Olympus IX71 microscope (Olympus Co.).
Expression of ACE2, eNOS, and iNOS Protein in the Lung
The expression of ACE2, eNOS, and iNOS in the lung of both WT and ACE2-/y mice was measured using Western blot analysis as previously described (27). Briefly, the left upper lobe of the lung was snap frozen in liquid nitrogen and later processed for lung homogenates in RIPA lysis buffer (Beyotime Institute of Biotechnology, Haimen, China) containing an antiprotease cocktail (1 mmol/L PMSF, 1 mmol/L NaF, and 1 mmol/L Na3VO4; US Biological Inc., Swampscott, MA, USA) to extract total proteins. Protein lysates were quantified using a BCA protein assay kit (Beyotime Institute of Biotechnology). The proteins were separated using 8% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Beyotime Institute of Biotechnology) and were transferred onto PVDF membranes (Millipore, Bedford, MA, USA). The membranes were blocked in Tris-1 buffer (Biosharp Biotechnology, Hefei, China) at pH 7.4 containing 0.1% Tween 20 (TBST; Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) and 5% bovine serum albumin (Roche, Ltd., Basel, Switzerland) for 1 h at room temperature and were then incubated with primary antibodies against ACE2 (1:100 dilution; Abcam Ltd.), iNOS (1:1,000 dilution; Cell Signaling Technology, Beverly, MA, USA), eNOS (1:1,000 dilution; Cell Signaling Technology), or β-actin (1:3,000 dilution; Bioworld technology, Co. Ltd.) at 4°C overnight. The blots were washed with TBST and incubated with goat anti-rabbit IgG conjugated with horseradish peroxidase (1:10,000 dilution; Zhongshan Golden Bridge Biotechnology Co. Ltd., Beijing, China) for 1 h at room temperature. Immunoreactive complexes were visualized using chemiluminescence reagents (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Quantification of ACE2 Protein in the Serum, Lung, Liver, and Spleen
Blood was obtained via aspiration after right heart puncture, and the serum was collected by centrifugation. The left lower lobe of the lung, the left lobe of the liver, and the spleen were homogenized and centrifuged at 93 × g for 5 min at 4°C to obtain the supernatants. The levels of ACE2 in the serum and organs were quantified using an ELISA kit (Shanghai Westang Bio-tech. Co., Ltd.) according to the manufacturer's instructions.
Cell Counts in the Bronchoalveolar Lavage Fluid
Bronchoalveolar lavage fluid (BALF) was collected according to a previous report (29) from the WT control, ALI, MSC-GFP, and MSC-ACE2 groups (n = 6 per group). The total cell counts were determined using a hemocytometer (Thermo Fisher Scientific, Inc.). The differential cell counts were measured using Wright's dye staining (Jiancheng Bioengineering Co., Nanjing, China). The number of neutrophils was calculated as the total number of cells multiplied by the percentage of neutrophils in the BALF samples. All analyses were performed in a blinded fashion.
Lung Histopathology
The lung sections from the right upper lobe were stained using a hematoxylin and eosin staining kit (Beyotime Institute of Biotechnology). The severity of the lung injury was quantified blindly according to a previously published scoring system (8) by a pathologist, based on the images of 10 randomly selected high-power fields (400×). For each section, following the scoring system, five criteria were included: edema, alveolar and interstitial inflammation, alveolar and interstitial hemorrhage, atelectasis, and hyaline membrane formation. Each criterion was graded according to a 4-point scale. A total lung injury score was calculated as the sum of the five criteria.
Measurement of Ang II, Ang 1–7, and Several Cytokines
The left lower lobe of the lung was snap frozen and later processed for lung homogenates. The concentration of Ang II and Ang 1–7 in the lung tissues was evaluated using an ELISA kit (Jiancheng Bioengineering Co.); the levels of IL-1β, IL-6, and IL-10 in the lung and the levels of IL-1β in the serum were measured using an ELISA kit (ExCellBio, Shanghai, China) strictly according to the manufacturer's instructions.
Evaluation of the Lung Edema
The lung edema of the control, ALI, MSC-GFP, and MSC-ACE2 groups of both the WT (n = 6 per group) and ACE2-/y mice (n = 3 per group) was measured using the lung wet weight/body weight ratio (LWW/BW), which reflects the microvascular endothelial permeability of the lung (8,22,26).
Measurement of Evans Blue Leakage
In the control, ALI, MSC-GFP, and MSC-ACE2 groups of WT mice (n = 5 per group), Evans blue (20 mg/ kg in 80 μl saline; Sigma-Aldrich) was injected into the tail vein. Thirty minutes later, the right ventricle of the heart was perfused with 10 ml of heparinized saline to flush the lungs of the intravascular dye. A complete perfusion was confirmed when all of the blood had been cleared from the lung. The right lung tissue (100 mg) was then incubated in formamide (Sigma-Aldrich) for 24 h at 60°C. The extracted Evans blue was measured using a spectrophotometer at 630 nm.
Ultrastructure of the Pulmonary Endothelial Cells
Small fragments (2 × 2 × 2 mm) of tissue were removed from the middle lobe of the right lung and fixed in 2.5% glutaraldehyde (Sigma-Aldrich) in 0.1 M phosphate buffer (pH = 7.4) and subsequently fixed in 1% osmium tetroxide (Sigma-Aldrich). The fragments were washed three times with PBS, dehydrated in a graded ethanol series, and embedded in araldite resin (Sigma-Aldrich) blocks. The samples were cut into 0.2-μm- to 2-μm-thick sections and stained with uranyl acetate (Amresco) and lead citrate (Sigma-Aldrich) for transmission electron microscope (JEM-1011; JEOL Ltd., Tokyo, Japan) analysis.
Statistical Analysis
All data are presented as the means ± standard deviations (SD). Statistical analyses were performed using SPSS 16.0 (IBM, Armonk, NY, USA). The comparison among groups was performed using one-way ANOVA followed by Bonferroni's post hoc test. Comparisons between two groups were made using an unpaired Student's t-test. Values of p < 0.05 were considered statistically significant.
Results
ACE2 Transduction of MSCs
The fluorescence microscopy (Fig. 1A) and flow cytometry images (Fig. 1B) showed the expression of eGFP. The transduction efficiency was 86–94% and was well maintained over 30 passages. The expression of ACE2 mRNA in the MSC-ACE2 group was approximately fourfold greater than that of the MSC group (Fig. 1C). ACE2 protein expression was also increased in the MSC-ACE2 group; the ACE2 transgene not only resulted in an approximately twofold increase in ACE2 protein levels in the transduced MSCs (Fig. 1D) but also resulted in an approximately threefold increase in the ACE2 secreted into the culture media (Fig. 1E). Therefore, ACE2 expression in transduced MSCs was approximately fivefold higher than that of nontransfected MSCs group.

Transduction efficiency and measurement of angiotensin-converting enzyme 2 (ACE2) mRNA and ACE2 protein after ACE2 gene transduction. (A) Phase control and fluorescence microscopy of mesenchymal stem cells (MSCs), MSC-GFP, and MSC-ACE2 at passages 4, 6, and 6, respectively (100×, scale bar: 50 μm). (B) Transduction efficiency was evaluated by detecting the expression of eGFP using flow cytometry and was as high as 94.3%. (C) Detection of ACE2 mRNA in MSCs using RT-PCR. The amount of ACE2 mRNA in the MSC-ACE2 group is approximately four times as high as that of the MSC group (n = 3; *p < 0.05 vs. MSC; †p < 0.05 vs. MSC-GFP). (D) ACE2 protein expression in MSCs was evaluated using Western blot analysis. The ACE2 protein was highly expressed in the MSC-ACE2 group (n = 3; *p < 0.001 vs. MSC; †p0.001 vs. MSC-GFP). (E) Secreted ACE2 protein in the culture media was detected using an ELISA. The results show that the transfected MSCs could secrete ACE2 protein and that the ACE2 levels in the culture media of the MSC-ACE2 group were approximately threefold greater than those of the nontransfected MSC group.
The Tracking and Retention of MSC-Derived Cells in Mice with LPS-Induced ALI
NIR fluorescence imaging of both the whole body in vivo and the organs ex vivo showed that the MSC-ACE2 cells were predominantly localized within the lung 30 min after their delivery and peaked at 24 h, subsequently decreasing gradually, but remaining high until 72 h; weak signals were also detected in the liver at 24 h and in the spleen at 72 h (Fig. 2A, B). The retention of MSCs in the lung was also verified by fluorescence micros copy at 72 h (Fig. 2C).

Detection of MSC-derived cells in the injured lungs. (A, B) NIR fluorescence imaging of both whole body in vivo and organs ex vivo showed that the MSC-ACE2 cells were predominantly localized within the lung 30 min after delivery. The signal in the lung peaked at 24 h and then decreased gradually, but remained high until 72 h; weak signals were also detected in the liver at 24 h and in the spleen at 72 h. The signals in the liver and spleen increased over the course of the experiment by 72 h (Fluo, fluorescence; C.C. fluo, color-coded fluorescence; n = 3 per group, *p < 0.05 vs. 30 min; †p < 0.05 vs. 24 h). (C) GFP+ cells (arrow) were detected by fluorescence microscopy 72 h after cell infusion via tail vein injection following the lipopolysaccharide (LPS) challenge (400×, scale bar: 20 μm).
The Enhanced Local Expression of ACE2 Protein in the Lungs
The expression of ACE2 protein decreased significantly due to the LPS-induced damage of the endothelium in the ALI group. At 24 and 72 h after the treatment with MSC-ACE2, the expression of ACE2 in the lung recovered significantly both in the WT and ACE2-/y mice (Fig. 3A, B), although the ACE2 level measured by both Western blotting (Fig. 3A) and ELISA (Fig. 3C) showed a decrease at 72 h compared to 24 h [Lung: 24 h (0.85 ± 0.09) vs. 72 h (0.75 ± 0.05) ng/mg protein, p = 0.036] (Fig. 3C). ACE2 levels in the serum at 72 h did not change after MSC-ACE2 treatment, and the lung-to-serum ratio of ACE2 was high in the MSC-ACE2 group (Fig. 3B). Interestingly, we also found that there was an increase in ACE2 protein in the liver and spleen at 72 h, and it was more significant in liver [liver: 24 h (0.46 ± 0.08) vs. 72 h (0.71 ± 0.08) ng/mg protein, p < 0.001; spleen: 24 h (0.41 ± 0.1) vs. 72 h (0.54 ± 0.1) ng/mg protein, p = 0.052] (Fig. 3C). However, the ACE2 levels in these organs were lower than that in the lung, suggesting that the majority MSC-ACE2 targeted the lung and locally overexpressed ACE2 protein.
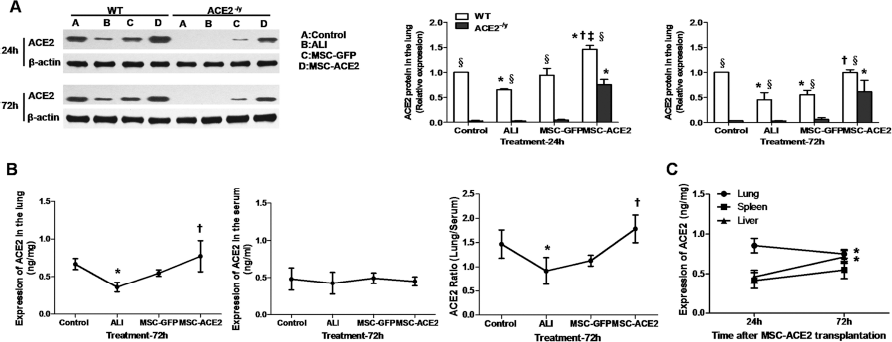
Effects of MSC-GFP and MSC-ACE2 injection on the expression of ACE2 in the lungs of mice. (A) ACE2 expression decreased significantly at 24 and 72 h in WT mice after LPS exposure and recovered after the treatment with MSC-ACE2. In the ACE2-/y mice, the expression of ACE2 was lost compared with that of the WT mice. There was no change after the LPS challenge, and only a slight increase in ACE2 protein was observed 72 h after the treatment with MSC-GFP. However, the expression of ACE2 increased greatly after MSC-ACE2 delivery, both in the WT and ACE2-/y mice; the increase in ACE2 protein was higher at 24 h than at 72 h. (B) The concentration of ACE2 protein in the lung tissue was initially reduced and recovered to normal levels after the MSC-ACE2 treatment, and there was no change in the serum of the four groups. The ACE2 ratio was calculated as the concentration of ACE2 in the lung over that in the serum of WT mice. (C) Expression of ACE2 in the lung, liver, and spleen of WT mice at 24 and 72 h following MSC-ACE2 transplantation. The expression of ACE2 in the lung was decreased at 72 h, while a corresponding increase in the liver and spleen was observed at 72 h (n = 3-6 per group; *p < 0.05 vs. control; †p < 0.05 vs. ALI group; ‡p < 0.05 vs. MSC-GFP group; §p < 0.05 vs. ACE2-/y group at the same time point).
MSC-ACE2 Delivery Decreased the Level of Ang II and Increased the Level of Ang 1–7 in Lung Tissues
LPS-treated WT and ACE2-/y mice showed significantly greater Ang II levels compared with control mice at 24 and 72 h, which decreased after MSC-GFP and MSC-ACE2 delivery. Differences were readily apparent between the MSC-ACE2 and MSC-GFP groups, and the expression of Ang II was higher in the ACE2-/y mice (Fig. 4A). Moreover, the results also showed that there was a corresponding increase in Ang 1–7 in the lung tissue of the MSC-ACE2 group at 24 and 72 h (Fig. 4B), which suggested that the increased ACE2 enzyme in the lung degraded the Ang II into Ang 1–7.

MSC-ACE2 transplantation degraded Ang II into Ang 1–7 in lung tissue. (A) LPS-treated WT and ACE2-/y mice showed significantly greater angiotensin (Ang) II levels compared with those of control mice at 24 and 72 h. Ang II levels decreased after MSC-GFP and MSC-ACE2 delivery. Differences were readily apparent between the MSC-ACE2 and MSC-GFP groups. The expression of Ang II was higher in ACE2-/y mice. (B) Ang 1–7, the degradation product of Ang II, was decreased after the LPS challenge and recovered at 24 and 72 h following MSC-ACE2 delivery (n = 3–6 per group; *p < 0.05 vs. control; †p < 0.05 vs. ALI group; ‡p < 0.05 vs. MSC-GFP group; §p < 0.05 vs. ACE2-/y group at the same time point).
MSC-ACE2 Further Improved Lung Histopathology When Compared with MSC-GFP Alone
Histopathology was performed to evaluate the severity of lung injury. The lung sections from LPS-challenged WT mice at 24 and 72 h showed a marked interalveolar septal thickening, extensive inflammatory infiltrates, and diffuse interstitial and alveolar edema. The LPS challenge induced lung injuries with a lung injury score of approximately 9.8 at 24 h and 13 at 72 h. In ACE2-/y mice, the loss of ACE2 expression resulted in more severe ALI when compared with the WT mice, displaying thicker interalveolar and septal walls, more serious interstitial and alveolar edema, and inflammatory infiltrates. These animals also had higher lung injury scores of 13 at 24 h and 15 at 72 h. The administration of MSC-GFP and MSC-ACE2 improved lung histopathology to different degrees. This improvement was more apparent in the MSC-ACE2 treatment group: the final lung injury score was 2.8 in the WT mice and 4.1 in the ACE2-/y mice at 24 h and 5.7 in the WT mice and 8.6 in the ACE2-/y mice at 72 h (Fig. 5).

Histological evaluation of the therapeutic potential of MSC-GFP and MSC-ACE2 in ALI mice. The H&E staining (200×, scale bar: 20 μm) of lung sections from LPS-challenged WT mice at 24 and 72 h showed marked interalveolar septal thickening, extensive inflammatory infiltrates, diffuse interstitial and alveolar edema, and severe interstitial hemorrhage. The lung injury score was approximately 9.8 at 24 h and 13 at 72 h. In ACE2-/y mice, the loss of ACE2 expression resulted in more severe acute lung injury, with a lung injury score of 13 at 24 h and 15 at 72 h. The administration of MSC-GFP and MSC-ACE2 improved the lung histopathology to different degrees. This improvement was more apparent in mice treated with MSC-ACE2. The final lung injury score was 2.8 in WT mice and 4.1 in ACE2-/y mice at 24 h and 5.7 in WT mice and 8.6 in ACE2-/y mice at 72 h (n = 3–6 per group; *p < 0.05 vs. control, †p < 0.05 vs. ALI group; ‡p < 0.05 vs. MSC-GFP group; §p < 0.05 vs. ACE2-/y group).
MSC-ACE2 Improved the Ability of MSC-GFP to Moderate LPS-Induced Lung and Systemic Inflammation
The total inflammatory cell and neutrophil counts in the BALF were determined using Wright's stain. The total inflammatory cell number counted in the BALF increased approximately 22-fold at 24 h and approximately 24-fold at 72 h following the administration of LPS; this increase was attributed to an increase in neutrophils (approximately 72–78% of the total cells in the ALI group). The treatment with MSC-GFP reduced the total cell and neutrophil counts in the BALF; however, the treatment with MSC-ACE2 further reduced the BALF cell counts (Fig. 6A).

The therapeutic potential of MSC-GFP alone or MSCs transduced with the ACE2 gene for LPS-induced lung inflammation in mice. (A) Total cell and neutrophil counts in bronchoalveolar lavage fluid (BALF) increased following LPS administration. Treatment with MSC-GFP alone reduced the total cell and neutrophil counts in the BALF. However, treatment with MSC-ACE2 further reduced the BALF cell counts. (B–D) The levels of IL-6 and IL-1β were elevated, and the level of IL-10 was reduced in WT and ACE2-/y mice in response to the LPS challenge. Treatment with MSC-GFP and MSC-ACE2 decreased the levels of the proinflammatory cytokines and increased the expression of IL-10. The effect was more dramatic in the MSC-ACE2 group at 24 and 72 h. (E) The expression of IL-1β in the serum of ALI mice was also dramatically increased at 72 h and decreased after the treatment with MSC-GFP; MSC-ACE2 could further reduce the level of IL-1β in the serum to almost baseline levels (n = 3–6 per group; *p < 0.05 vs. control, †p < 0.05 vs. ALI group; ‡p < 0.05 vs. MSC-GFP group; §p < 0.05 vs. ACE2-/y group).
The levels of the proinflammatory cytokines IL-6 and IL-1β and the anti-inflammatory cytokine IL-10 were also measured in the lung homogenates. The expression of IL-6 and IL-1β were both elevated, while the level of IL-10 was reduced in the WT and ACE2-/y mice in response to the LPS challenge. The treatment with MSC-ACE2 decreased IL-6 and IL-1β levels and increased the expression of IL-10 to a greater extent than did the MSC-GFP group at both 24 and 72 h (Fig. 6B–D). While the level of IL-1β in the serum increased greatly 72 h after the LPS stimulation and decreased after the treatment with MSC-GFP, MSC-ACE2 delivery could further reduce the IL-1β concentration when compared with the MSC-GFP group (Fig. 6E).
MSC-ACE2 Improved the Function of MSC-GFP to Preserve Pulmonary Endothelial Functions
The expression of iNOS and eNOS in the lungs was examined by Western blotting to determine the synthesis functions of the endothelium. As illustrated in Figure 7, LPS stimulation increased iNOS protein expression and decreased eNOS protein expression at 24 and 72 h. Treatment with either MSC-GFP or MSC-ACE2 attenuated iNOS expression and increased eNOS expression at 24 and 72 h in the ALI mice. There were no significant differences between the WT and ACE2-/y mice in all of the groups (Fig. 7). In addition, the MSC-ACE2 treatment moderated the increase in lung edema, shown as LWW/BW (Fig. 8A), as well as the increase in lung endothelial permeability, as demonstrated by Evans blue extravasation (Fig. 8B), compared to the MSC-GFP group at 24 and 72 h after the LPS challenge in both the WT and ACE2-/y mice. The results of the pulmonary endothelium ultrastructure also confirmed the presence of injured pulmonary vascular endothelial cells in the ALI group at 72 h, as demonstrated by the disruption of endothelial cell–cell contacts, the presence of swollen endothelial cells, and the degeneration of cellular organelles. Interestingly, the MSC-GFP treatment showed a partial improvement against endothelial damage, while endothelial damage was nearly normalized in the MSC-ACE2 group (Fig. 8C).

Effects of MSC-GFP and MSC-ACE2 injection on the expression of iNOS and eNOS in the mouse lung. LPS stimulation increased iNOS protein expression and decreased eNOS protein expression at 24 and 72 h. Treatment with MSC-GFP and MSC-ACE2 attenuated iNOS expression and increased eNOS expression. There was no difference in iNOS protein expression between the MSC-GFP group and the MSC-ACE2 group. However, MSC-ACE2 further increased the expression of eNOS protein to the baseline values. There were no differences between WT and ACE2-/y mice in all groups (n = 3–6 per group; *p < 0.05 vs. control, †p < 0.05 vs. ALI group; ‡p < 0.05 vs. MSC-GFP group; §p < 0.05 vs. ACE2-/y group).

Effects of MSC-GFP and MSC-ACE2 injection on pulmonary vascular permeability. (A) A comparison of the wet lung-to-body weight ratio in different groups at 24 and 72 h (n = 6 per group) showed that MSC-ACE2 attenuated the lung edema more notably than MSC-GFP. (B) Representative images of Evans blue-injected lungs from WT mice 24 and 72 h after cell delivery. The dye accumulation in the lung increased dramatically at 24 and 72 h in the ALI group and was reduced by MSC-GFP and MSC-ACE2 treatment at 24 and 72 h. The dye leakage was normalized to baseline in the MSC-ACE2 group (n = 5 per group; *p < 0.05 vs. control, †p < 0.05 vs. ALI group, ‡p < 0.05 vs. MSC-GFP group). (C) A representative ultrastructural image from WT and ACE2-/y male mice at 72 h showing large openings in cell–cell contacts (arrow) in the endothelium, swelling of the cytoplasm, and the degeneration of cellular organelles in the ALI group. The MSC-GFP treatment showed a partial improvement against endothelial damage, while the endothelial damage was nearly normalized in the MSC-ACE2 group (frame represents a cell–cell junction). Scale bar: 0.2 μm.
Discussion
The present study suggests a further benefit of MSC-ACE2, when compared with MSC-GFP, in the treatment of ALI. The underlying mechanisms of the protective effects of MSC-ACE2 may be attributed to the inhibition of the inflammatory response and to the improvement of lung endothelial function. Namely, the MSC-ACE2 treatment 1) improved histopathological morphology; 2) further decreased the neutrophil counts in the BALF, downregulated the expression levels of the proinflammatory mediators IL-1β and IL-6, and upregulated the levels of the anti-inflammatory cytokine IL-10 in the lung tissue; and 3) significantly reduced lung edema, in part by improving lung endothelial permeability. We also found that treatment with MSC-ACE2 normalizes the expression of iNOS and eNOS in the lung. These further beneficial effects of MSC-ACE2 were also confirmed in the ACE2-/y mice.
These additional benefits, when compared with MSCs alone, are attributed to the overexpression of ACE2 protein in the injured lung after MSC-ACE2 transplantation. Our data are consistent with recent findings showing that recombinant human ACE2 could attenuate acid aspiration, LPS, peritoneal sepsis, and bleomycin-induced lung injuries (17,32). Current data show that ACE2 degrades Ang II into Ang 1–7. Interestingly, in this study, the increase in ACE2 expression in the lung after MSC-ACE2 delivery was correlated with a dramatic decrease in the levels of Ang II in the lung tissue, while the degradation product of Ang II, Ang 1–7, showed a corresponding increase. It appears that the overexpression of ACE2 in the lung via MSC-ACE2 transplantation possesses the enzymatic activity to cleave the lung-generated Ang II into Ang 1–7. Because systemic delivery of MSC-ACE2 may also result in an increase in ACE2 in the serum that could influence the concentration of Ang II in the lungs, we measured the serum ACE2 levels and found that MSC-ACE2 transplantation enhanced the expression of ACE2 in the injured lung without changing ACE2 levels in the serum. This result suggests that the downregulation of Ang II is mainly attributed to the overexpression of biologically active ACE2 protein in the lungs. This result also implies that the additional therapeutic effects of MSC-ACE2, compared to MSC-GFP, occurs mainly by reducing Ang II levels and by alleviating the deleterious effects of Ang II by the overexpression of ACE2 protein in the injured lung.
We also examined the localization of MSC-ACE2 to evaluate whether the overexpression of ACE2 protein in the lungs and the observed benefits were associated with the targeted retention of MSC-ACE2 in the injured lung. We found that a large proportion of the cells were localized in the injured lung 30 min after MSC-ACE2 delivery and peaked at 24 h, subsequently decreasing gradually, but remaining high until 72 h. Our results are consistent with previous studies in that the MSCs were mainly taken up by the injured lung 4 to 7 days postinjection (1,43,47). However, other studies have reported that there was only a low level of MSC retention in the injured lung (4,29). Herzog et al.'s study (13) showed that the localization of MSCs was related to the severity of the injury. Based on the histopathologic examination, the lung injuries induced in this study were more severe than those of a previous study in which there was a low level of MSC retention and, therefore, resulted in a higher uptake of MSCs in the injured lung that locally overexpressed ACE2 protein. Furthermore, it was shown that while MSC-ACE2 localization in the lung tissue was decreased at 72 h, it remained at a higher level than at 30 min after the cell transplantation and was correlated to an increase in cell localization in the liver and spleen. The expression of ACE2 in the lung also decreased at 72 h and correspondingly increased in the liver and spleen. Nonetheless, the transplantation of MSCs had only a minor effect on the liver and spleen, as the ACE2 levels in these organs were lower than that of the lung. Furthermore, the levels of ACE2 in the serum were not significantly changed after MSC transplantation. These findings strongly suggest that MSCs could deliver the ACE2 enzyme to the injured lung after cell transplantation and that the beneficial effects of these genetically engineered MSCs to further promote lung repair in ALI mice, when compared with MSCs alone, relied mainly on this targeted approach.
As shown in our study, the ACE2-modified MSCs could overexpress and secrete ACE2 locally in the lung after delivery and showed protective effects within a short time period. It is suggested that MSC-ACE2 exerts its therapeutic effects to promote lung repair mainly through a paracrine secretion function and that the shortterm retention of MSC-ACE2 is very important for this targeted therapy. MSC-ACE2 localization in the lung tissue was decreased at 72 h, suggesting that MSC-ACE2 cells were not likely engrafted into the lung. Furthermore, many other studies have also suggested that the engraftment and differentiation of MSCs is very low (4,29). These results suggest that the engraftment of MSC-ACE2 in the lung and subsequent differentiation into endothelial and epithelial cells that promote lung repair in this model of ALI was not the chief mechanism in this study.
In our study, utilizing ACE2 as a protective transgene was a critical strategy for ALI therapy and was based on the pathogenic mechanisms of ALI. Current evidence shows that the RAS, especially its main biological effector, Ang II, plays a critical role in the pathogenesis of ALI (7,15,16). In addition to regulating blood pressure, aldosterone release, and sodium reabsorption (3), recent studies have shown that Ang II is a strong proinflammatory mediator involved in the process of ALI/ARDS through the AT1R (16,17,23,27,45). In ALI animal models and ALI patients, the levels of Ang II in lung tissue and in circulation increased greatly (27,34). This increase was also observed in our study. In a previous study, we reported that the systemic infusion of Ang II into normal rats activated an inflammatory reaction and increased the endothelial permeability in the lung and caused an ALI; these detrimental effects could be inhibited by the AT1R antagonist losartan (48). Therefore, the downregulation of Ang II is an extremely important target to attenuate ALI.
The elevated level of Ang II in ALI is preceded by increased ACE expression and decreased ACE2 levels in lung tissue. Ang II is generated from Ang I by ACE (16,17,23,27,45), and a study by Wösten-van Asperen and colleagues (41) found that ACE activity was enhanced in BALF of ventilated LPS-exposed animals, whereas the activity of ACE2, a counterregulatory enzyme of ACE that degrades Ang II into Ang 1–7, was reduced. Imai et al.'s study (17), in addition to the results of the present study, also demonstrated that the expression of ACE2 was significantly decreased in ALI mice and that Ang II accumulation was increased. These effects were more significant in the ACE2-/y mice, and the loss of ACE2 resulted in a more severe lung injury. Currently, ACE inhibitors (30), an AT1R antagonist (27,45) and recombinant ACE2 (17) have been used in preclinical ALI animal models to downregulate the concentration of Ang II. ACE2 protein shows superiority when compared with ACE inhibitors and the AT1R antagonist, as ACE2 inactivates Ang II directly and its degradation product, Ang 1–7, also has a protective role in lung injury repair. In contrast, ACE inhibitors cannot eliminate the harmful effects produced by Ang II, and the AT1R antagonist cannot restore ACE2 expression (16). Based on these concepts, in the present study we delivered ACE2 enzyme to injured lung using genetically modified MSCs and found that the increased ACE2 could further improve the lung histopathology of both WT and ACE2-/y mice at 24 and 72 h when compared with the MSC-GFP group. In addition, MSC-ACE2 significantly improved the ability of MSC-GFP to moderate the LPS-induced inflammatory response and to preserve pulmonary endothelial function. These results showed that ACE2-transfected MSCs possess significant additional protective effects when compared with MSCs alone, which provides further evidence that ACE2 is a suitable target for overexpression in the treatment of ALI.
The delivery of a protective gene using MSCs could overcome the limitations of transient gene expression, host immunoinflammatory responses, and unspecific cell targeting by classic viral or nonviral vectors because MSCs have been suggested to have low immunogenicity and a targeting bias toward injured sites. These properties make MSCs an ideal gene vector to carry the ACE2 gene to the injured site and to permit sustained over-expression of ACE2 locally in the lung. In the present study, MSC-based ACE2 gene therapy is shown to be a feasible therapeutic approach for treating ALI. First, combining MSCs with the ACE2 gene provides additional benefits in attenuating lung injury. The results of our study revealed that ACE2-transfected MSCs elicited further improvement of lung injury when compared with MSCs alone. Second, MSCs have been suggested to be “immune privileged” or protected from host rejection (19,20). The modification of MSCs with a viral vector seems to have no major influence on the immunosuppressive properties of MSCs in our study because the inflammation in the injured lung was inhibited by MSC-ACE2 transplantation; this result is consistent with another study that has shown that no systemic inflammatory responses were invoked (36). Additionally, our study found that MSC recruitment to the injured lung facilitated selective and durable ACE2 expression in the lung, and the increased ACE2 enzyme possessed the bioactivity to degrade Ang II into Ang 1–7, which also facilitated local repair. These results indicate that MSCs are a good ACE2 gene vector and that the combination of MSCs and the ACE2 gene enhanced the therapeutic effects of MSCs.
In this study, MSC-GFP transplantation also showed some benefits in the treatment of ALI mice. This result is reasonable, as a number of previous studies have reported that bone marrow progenitor cells and freshly isolated human cord blood CD34+ progenitor cells could inhibit lung inflammation, restore the integrity of lung vasculature, attenuate the severity of lung injury, and improve the mortality of ALI mice following an LPS challenge (14,46). Given that MSC-GFP cells could only express and secrete low levels of ACE2 protein, the beneficial effects of MSCs in lung repair may not be mainly through the ACE2 protein; other mechanisms may be involved in promoting lung repair. Because the engraftment of MSCs to the lung is low, it is currently believed that MSCs exert their therapeutic effects mainly through paracrine mechanisms. MSCs possess the ability to secrete multiple soluble factors, such as growth factors, anti-inflammatory cytokines, and antimicrobial peptides. These factors can regulate endothelial and epithelial permeability, modulate innate and adaptive immunity, improve alveolar fluid clearance, and so on (39).
The main limitation of this study is its focus on the short-term (24 h to 72 h) therapeutic effects of MSC-ACE2 treatment. Data on the long-term effects are lacking and will require further study. In addition, our study has shown that there was also an increase in ACE2 enzyme expression in the liver and spleen at 72 h, which was correlated with an increase in MSC localization to these organs. A study by Osterreicher and colleagues (31) has suggested that the administration of recombinant ACE2 inhibits liver fibrosis in murine models of liver injury. Whether the increased ACE2 levels in the liver and spleen have beneficial effects in these two organs were not investigated in this study, and further work is still needed. Another limitation is that the LPS-induced model of ALI does not fully recapitulate the features of human lung injury (5). These findings should be reproduced in more clinically relevant models, such as animal models of pneumonia and sepsis-induced ALI.
Conclusion
In conclusion, MSCs modified with the ACE2 gene localized to the injured lung and provided an additional attenuation of the lung inflammatory response and enhanced pulmonary endothelial function compared with MSCs alone. This effect occurred mainly through the reduction of Ang II expression in the lung due to the local overexpression of the ACE2 protein. These results suggest a novel strategy for the targeted treatment of LPS-induced ALI. MSC-based ACE2 gene therapy represents a new hope for patients with ALI.
Footnotes
Acknowledgments
We thank Jun Liu, Liang Dong, and Yanli An for their technical assistance. This work was supported by the National Natural Science Foundation of China under the contract grant Nos. 81000828, 81170057, 81201489, and 81372093, and the Natural Science Foundation of Jiangsu Province under the contract grant No. BK20131302. This work was also supported by the Graduate Innovation Project of Jiangsu Province under the contract grant Nos. CXLX_0151, CXLX13_123. The authors declare no conflict of interest.