
Abstract
We have previously developed a robust regimen for tolerance induction in murine models of islet cell transplantation using pre- and posttransplant infusions of donor splenocytes (SPs) treated with a chemical cross-linker ethylcarbodiimide (ECDI). However, the requirement for large numbers of fresh donor SPs for ECDI coupling impairs its clinical feasibility, and additionally, the compatibility of this tolerance regimen with commonly used immunosuppressive drugs is largely unknown. In the current study, we demonstrate that equivalent tolerance efficacy for islet cell transplantation can be successfully achieved not only with a significantly lower dose of ECDI-SPs than originally established but also with culture-expanded donor B-cells or with soluble donor antigens in the form of donor cell lysate, which is ECDI coupled to recipient SPs. We further demonstrate that tolerance induced by donor ECDI-SPs is dependent on a favorable apoptotic-to-necrotic cell ratio post-ECDI coupling and is not affected by a transient course of conventional immunosuppressive drugs including tacrolimus and mycophenolate mofetil. While splenic antigen-presenting cells of the recipient play an important role in mediating the tolerogenic effects of donor ECDI-SPs, splenectomized recipients can be readily tolerized and appear to employ liver Kupffer cells for uptaking and processing of the ECDI-SPs. We conclude that infusion of donor ECDI-SPs is a versatile tolerance strategy that has a high potential for adaptation to clinically feasible regimens for tolerance trials for human islet cell transplantation.
Keywords
Introduction
We have previously developed a robust regimen for induction of donor-specific transplantation tolerance using pre- and posttransplant infusions of donor splenocytes (SPs) treated with a chemical cross-linker 1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide (ECDI-SPs) (13,16). This strategy was initially established in murine allogeneic islet cell transplant models, in which tolerance can be effectively induced to islet allografts transplanted via several different routes, including the kidney capsule, intraportal infusion, which is currently in clinical practice, as well as bioengineered scaffolds implanted in the abdominal fat pad (13,16). This tolerance strategy is also shown to be highly efficacious in a murine allogeneic heart transplant model (2) and in murine xenogeneic islet transplant models (30) and is now being tested in nonhuman primates for tolerance induction to islet allografts and islet xenografts. Mechanistic studies reveal that tolerance by this protocol is mediated through targeting recipient splenic antigen-presenting cells (APCs) and a PD-1/PD-L1-dependent downregulation of effector T-cell activity as well as expansion of regulatory T-cells (12,16).
Moving forward to preclinical nonhuman primate tolerance studies and ultimately to human islet cell transplantation tolerance trials, several critical issues of this strategy pertaining to its clinical applicability must be addressed. First, a cell dose calculation based on body weight suggests that 4 × 109 cells/kg/infusion was the dose successful in rodent models. However, obtaining such large numbers of fresh donor SPs for ECDI coupling and infusions will be difficult to achieve in nonhuman primates or in humans (18). Therefore, establishing the minimal cell number required for tolerance induction will provide the basis for dose-finding experiments in large animals and eventually in human trials. Furthermore, identifying alternative and/or expandable sources of donor cells for ECDI coupling will significantly alleviate the need for procuring fresh cells directly from the donor, thereby extending the potential applicability of this approach to both live and deceased organ donor transplantation. Our previous data suggest that the vast majority (>90%) of the infused ECDI-fixed donor cells are rapidly internalized by recipient splenic phagocytes, which then process and present the donor antigens in the context of recipient major histocompatibility complexes (MHCs) (12). This observation raises the intriguing question of whether tolerance can be induced with soluble donor antigens delivered to the recipient phagocytes without having to use intact donor cells. It also raises the question of whether the presence of a spleen is mandatory for such tolerogenic interactions. Lastly, for appropriate design of clinical trials to test this tolerance strategy in human transplantation, its compatibility with conventional immunosuppressive drugs must be determined (14,15).
In the current study, we used a murine allogeneic islet cell transplant model to address these issues. Our data suggest that tolerance can be effectively achieved with not only considerably fewer donor cells but also with soluble donor antigens delivered in the form of donor cell lysate that is ECDI coupled to recipient cells as carriers. Furthermore, absence of the spleen does not affect tolerance induction by donor ECDI-SPs; rather hepatic APCs assume the role of interacting with and phagocytosing the infused ECDI-SPs. Lastly, standard immunosuppressive drugs, including tacrolimus (FK) and mycophenolate mofetil (MMF), do not impair tolerance efficacy by ECDI-SPs. These data thus provide important parameters for translating this strategy to large animal experiments and ultimately to the design of clinical trials for human islet cell transplantation.
Materials and Methods
Mice
Eight- to 10-week-old male BALB/c, C57BL/6 (B6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All mice were housed under specific pathogen-free conditions at Northwestern University (NU, Chicago, IL, USA). All studies were approved by the NU Institutional Animal Care and Use Committee. Male cluster of differentiation 47 (CD47) knockout mice (B6 background) spleens were kindly provided by Dr. Yong-guang Yang (Columbia Center for Translational Immunology, Columbia University, New York, NY, USA).
Diabetes Experiments
For diabetes experiments, mice were treated with streptozotocin (Sigma-Aldrich, St. Louis, MO, USA) at 200 mg/kg (IP injection). Two consecutive glucose readings >250 mg/dl measured from tail bleeding using OneTouch Ultra glucometer (LifeScan, Inc., Milpitas, CA, USA) were used to diagnose diabetes. Islet isolation and transplantation have been described previously (17). Approximately 500 islets were implanted under the left kidney capsule of recipient mice. Graft rejection was determined by two consecutive blood glucose readings >250 mg/dl. For the splenectomy procedure, a small incision was made on the exposed abdominal wall to provide access to the spleen. The spleen was then pulled out through the incision, and a 4-0 silk suture (Ethicon, Somerville, NJ, USA) was used to ligate the splenic blood vessels. The spleen was then removed by transecting the blood vessels distal to the ligature. The skin incision was then closed using 5-0 a silk suture (Ethicon). FK (Astellas Pharma, Northbrook, IL, USA) was dissolved in normal saline and injected at 3 mg/kg/day subcutaneously. MMF (Roche, Madison, WI, USA) was dissolved in normal saline and injected at 100 mg/kg/day IP. The drugs were given 1 day prior to transplant and continued to 8 days posttransplant.
ECDI Cell Coupling and Tolerance Induction
Tolerance was induced by IV injection of ECDI-treated donor SPs. Briefly, SPs were incubated with ECDI (Calbiochem, Torrey Pines, CA, USA) [every 3.2 × 108 cells in 1 ml of Dulbeccos' phosphate-buffered saline (DPBS; Life Technologies, Grand Island, NY, USA) with a final concentration of 30 mg/ml of ECDI] on ice for 1 h with agitation on an agitator (Labline Instruments, Inc., Melrose Park, IL, USA) followed by washing and injection into the recipients via tail vein at the indicated doses on the indicated days. Alternatively, mouse SPs were frozen with freezing medium [10% dimethyl sulfoxide (Sigma-Aldrich) +90% fetal bovine serum (Gibco, Burlington, ON, Canada)] and kept in liquid nitrogen. For thawing, frozen SPs were transferred to a 37°C water bath and gently agitated until thawed. Cells were then washed with DPBS three times prior to ECDI coupling. Donor cell lysate was prepared by three cycles of freeze—thaw of SPs and incubated with equal numbers of recipient SPs in the presence of an identical concentration of ECDI and washed as above. For tracking of ECDI-treated SPs in vivo, ECDI-fixed SPs were further labeled with PKH-67 (Sigma-Aldrich) at a final concentration of 2 × 10−6 M at 1 × 107 cells/ml at room temperature for 5 min, followed by washing prior to injection. The A20 B-cell lymphoma cell line was purchased from the American Type Culture Collection (Manassas, VA, USA), and seeded at 106/ml Roswell Park Memorial Institute 1640 (Gibco) supplemented with 10% heat-inactivated fetal bovine serum and grown at 37°C and 5% CO2. Cells were grown until confluent and split at a 1:10 dilution every 5—7 days. Harvested A20 cells were ECDI coupled as described above and injected at 5 × 107cells per mouse in a final volume of 300 μl IV.
FACS Analysis
To determine the percentage of apoptosis and necrosis of SPs after ECDI coupling, the flow cytometric Annexin-V plus propidium iodide (PI) assay (BD-Pharmingen, San Diego, CA, USA) was used. The cells were stained with annexin V and PI according to the manufacturer's instructions, followed by immediate fluorescence-activated cell sorting analysis. The percentages of annexin V+PI- (early stage apoptosis) cells, annexin V+PI+ (late stage apoptosis/early necrosis) cells, and annexin V-PI+ (necrosis) cells were recorded. Total necrosis was determined by the percentage of total PI+ cells calculated as: % annexin V+PI+ + % Annexin V-PI+. Liver APCs were isolated from C57BL/6 mice by perfusion of the liver with 0.5 mg/ml collagenase (Roche) in Hank's buffered salt solution (HBSS; Life Technologies). Fragmented livers were incubated for 20 min in digestion media (the 0.5 mg/ml collagenase in HBSS) at 37°C in an agitating incubator, passed through a nylon mesh (BD Falcon™, Franklin Lakes, NJ, USA), and stained with phycoerythrin (PE)-conjugated anti-IAb [AF6 (250 μg/ml); BD Biosciences, San Jose CA, USA], peridininchlorophyll protein complex-conjugated anti-F4/80 (BM8, 0.5 mg/ml), allophycocyanin-conjugated anti-CD11b (M1/70, 0.5 mg/ml), both from eBioscience (San Diego, CA, USA). For staining of A20 cells, fluorescein isothiocyanate-conjugated anti-H-2Kd [Clone SF1-1.1 (0.5 mg/ml); BD Biosciences] and PE-conjugated anti-I-Ad [clone AMS-32.1 (0.2 mg/ml); BD Biosciences] were used.
Graft Histology and Immunofluorescence
Kidneys bearing islet grafts were snap frozen in OCT compound (Sakura Finetek USA, Inc., Torrance, CA, USA) with liquid nitrogen and sectioned at 4 μm thickness. For histology, tissue samples were stained with hematoxylin and eosin (Sigma-Aldrich) and analyzed using Zeiss Axioskoplight microscopy (Thornwood, NY, USA). For visualization of CD4 and CD8 T-cells, sections were stained with rat anti-mouse CD4 mAb (1:250, rat IgG2a, k clone H129.19; BD Biosciences), or rat anti-mouse CD8 (1:250, rat IgG2a, k clone 53-6.7; BD Biosciences), followed by biotinylated donkey anti-rat (1:250) and cyanine 3 (Cy3)-streptavidin (BD Biosciences). For visualization of insulin, sections were stained with guinea pig anti-insulin polyclonal antibody (1:200, #A0564; Dako, Carpinteria, CA, USA) followed by visualization with donkey anti-guinea pig Alexa 488 (1:250; Jackson ImmunoResearch, West Grove, PA, USA). A 4′,6-diamidino-2-phenylindole (DAPI) stain (Life Technologies) was performed to identify cell nuclei. All negative controls were performed by eliminating the primary antibodies. Images were visualized using Zeiss Axio Scope A1, acquired with Jenoptik ProgRes MFcool camera (Jenoptik, Jena, Germany) and analyzed with ProgRes Mac Capture Pro 2.7 software (Jenoptik).
Statistical Analysis
Graft survival was calculated by Kaplan—Meier analysis. Log rank test was used to compare survival between groups. A Student t-test was applied to compare % of PI+ cells, % of apoptosis, and % PKH+ Kupffer cells. Values of p < 0.05 were considered to be statistically significant.
Results
The Dose Response of Donor ECDI-SPs Required for Tolerance Induction for Allogeneic Islet Cell Transplantation
We have previously shown that infusions of 1 × 108 donor ECDI-SPs on both day −7 and day +1, with day 0 being the day of transplantation, provide indefinite islet allograft survival in a full MHC-mismatched BALB/c to B6 (H-2d to H-2b) islet transplant model (16). We performed a dose titration to define the minimal dose of donor ECDI-SPs needed for the observed graft protection. As shown in Figure 1A, for each of the day −7 and day +1 infusion, 1 × 107 donor ECDI-SPs or above provided similar graft protection as the 1 × 108 dose. However, further decreasing of the dose to 5 × 106 cells per infusion significantly compromised the graft protection induced by donor ECDI-SPs. We conclude that for allogeneic islet transplantation, uncompromised allograft protection can likely be achieved at approximately one tenth of the dose of donor ECDI-SPs previously used. This finding significantly enhances the feasibility of donor ECDI-SPs in clinically relevant settings.
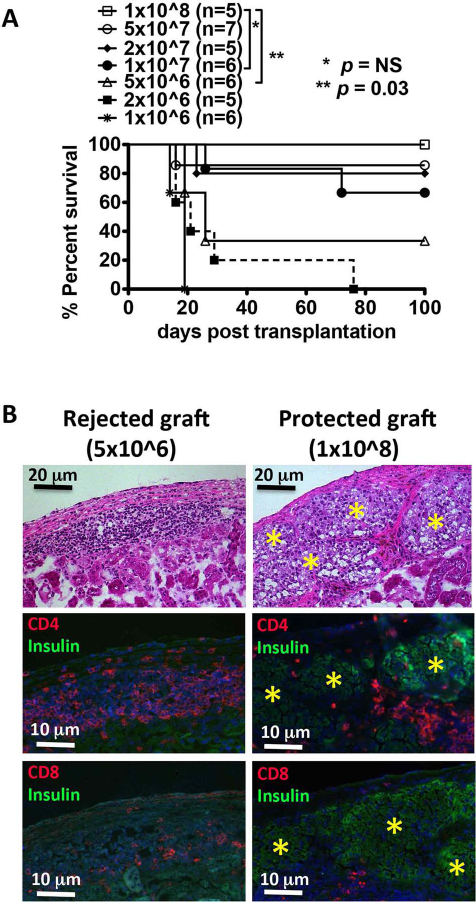
The dose response of donor ECDI-SPs for tolerance induction for allogeneic islet cell transplantation. (A) ECDI-fixed BALB/c SPs (ECDI-SPs), 1 × 106 to 1 × 108, were infused to diabetic B6 recipients on day −7 and day +1. BALB/c islets were transplanted on day 0. Blood glucose (BG) levels were followed until rejection occurred (BG > 250 mg/dl on 2 consecutive days) or 100 days posttransplantation, whichever came first. *p = 0.176, 1 × 108 versus 1 × 107; **p = 0.03, 1 × 108 versus 5 × 106. (B) Representative histological examination of rejected grafts (from recipients treated with 5 × 106 ECDI-SPs) and protected grafts (from recipients treated with 1 × 108 ECDI-SPs) retrieved on day 20 posttransplantation.*Discernable islets. Top: H&E (magnification: 10×); middle: triple immunofluorescent staining with insulin, CD4 and DAPI (magnification: 20×); bottom: triple immunofluorescent staining with insulin, CD8 and DAPI (magnification: 20×). Histology is representative of three each islet allografts obtained and sectioned from recipients treated with either the reduced dose (5 × 106) or the full dose (1 × 108) of ECDI-SPs.
We performed a histological examination of the rejected grafts at a low dose (5 × 106) of ECDI-SPs in comparison to the protected grafts at the high dose (1 × 108) of ECDI-SPs, both retrieved from the recipients around day 20 posttransplantation. As shown representatively in Figure 1B, the rejected grafts showed dense lymphocytic infiltration of predominantly CD4+ T-cells, but also a few CD8+ T-cells in the graft, with no visible islet structure or insulin staining. Conversely, the protected grafts showed well-preserved islet structure and positive insulin staining, with markedly decreased and often peri-islet instead of intraislet lymphocytic infiltration of CD4+ or CD8+ T-cells. Therefore, rejection in recipients treated with suboptimal doses of ECDI-SPs appears to be driven by an incomplete control of alloreactive T-cell responses as we have previously established (12).
Frozen Donor SPs Have Compromised Ability to Induce Transplant Tolerance Compared with Fresh Donor SPs for ECDI Coupling
Given that multiple doses of donor ECDI-SPs may be necessary for transplant tolerance induction (2), the clinical applicability of this approach would be significantly enhanced if stored frozen donor cells could be used for ECDI coupling and recipient infusions, particularly in a setting of deceased donor transplantation. We next tested whether frozen donor SPs could be used for ECDI coupling for transplant tolerance induction in the BALB/c to B6 islet transplant model. BALB/c SPs were collected and frozen with freezing medium for at least 24 h prior to slow thawing and ECDI coupling as described in the Materials and Methods. Two infusions of 108 donor ECDI-SPs prepared from thawed BALB/c SPs were given to B6 recipients on day −7 and day +1, with day 0 being the day of BALB/c islet transplantation. As shown in Figure 2A, donor ECDI-SPs prepared from frozen/thawed donor SPs only provided a marginal protection to the islet allografts. We hypothesized that frozen/thawed donor SPs may contain a high percentage of necrotic cells, thereby negating the tolerogenic effect of apoptotic ECDI-SPs seen when fresh donor SPs are used. Indeed, as shown in Figure 2B and C, frozen/thawed donor SPs after ECDI coupling showed a reversed ratio of necrotic to apoptotic cells compared with fresh donor SPs after ECDI coupling at 2, 4, and 6 h of culture at 37°C. Therefore, frozen/thawed donor cells with a high percentage of necrotic cells appear to be not ideal for ECDI coupling and tolerance induction.
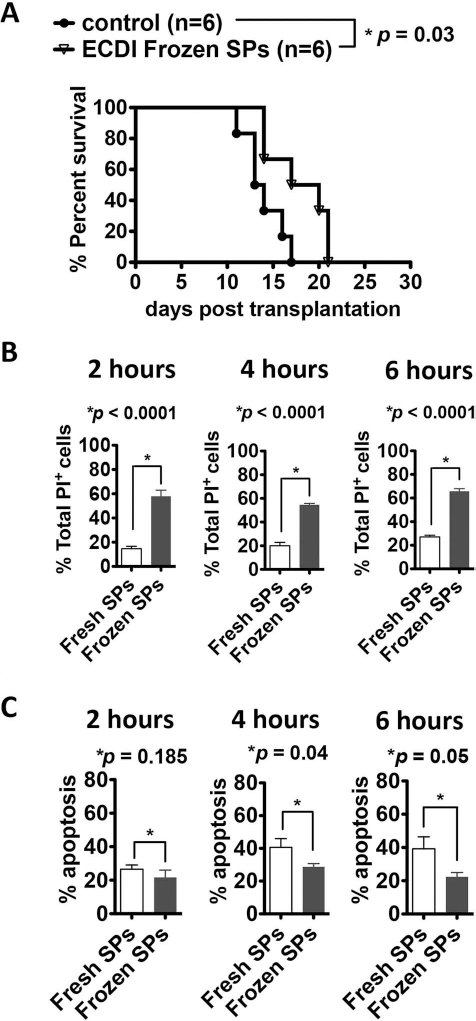
Frozen donor SPs have compromised ability to induce transplant tolerance compared with fresh donor SPs for ECDI coupling. (A) BALB/c ECDI-SPs prepared with frozen/thawed BALB/c SPs were injected on day −7 and day +1, and BALB/c islets were transplanted on day 0. Graft survival was monitored by serial BG levels. Control = untreated B6 recipients. *p = 0.03, frozen/thawed BALB/c ECDI-SPs versus control. (B, C) Percentages of necrotic (B) and apoptotic (C) cells at 2, 4, and 6 h of culture at 37°C after ECDI coupling. Annexin V+PI+ cells and annexin V-PI+ cells combined were counted as necrotic cells. Annexin V+PI- cells were counted as apoptotic cells. Comparisons were made between fresh versus frozen/thawed SPs. Results shown for (B) and (C) are the average from four independent experiments.
Donor Cell Lysate Coupled to Recipient SPs Via ECDI Is Equally Tolerogenic as Intact Donor Cells That Are Directly ECDI Fixed
To test if other storable forms of donor antigens can be used for ECDI-fixed cell-based tolerance strategy, we tested donor (BALB/c) cell lysates as a soluble form of donor antigens. BALB/c cell lysate was prepared by three cycles of rapid freezing/thawing of BALB/c SPs. The resulting donor cell lysate was then coupled to recipient (B6) SPs via the same ECDI coupling reaction. We reasoned that this method mimics peptide-coupled autologous SPs used for tolerance induction to peptide autoantigens in autoimmune disease models such as autoimmune diabetes and autoimmune demyelinating disease and therefore should similarly allow tolerance induction to the fixed soluble donor antigens. A dose of 1 × 108 B6 SPs ECDI coupled with BALB/c cell lysate were injected to diabetic B6 recipients on day −7 and day +1. BALB/c islet grafts were transplanted on day 0. As shown in Figure 3A, islet allografts in recipients treated with this regimen were equally protected as those in recipients treated with directly ECDI-fixed BALB/c SPs. Interestingly, while our previous data showed that donor ECDI-SPs are able to target both the direct and the indirect pathways of allorecognition (12), data in Figure 3A suggest that targeting the indirect allorecognition pathway alone by the ECDI-fixed cell strategy may be sufficient for tolerance induction in the allogeneic islet transplant model and is therefore likely the main contributor to graft protection by this method. We conclude that donor cell lysate as a storable form of donor antigens can be coupled to recipient SPs via ECDI for effective donor-specific tolerance induction.

Alternative sources of donor antigens for ECDI coupling and tolerance induction. (A) Infusions of 1 × 108 B6 SPs ECDI-fixed with BALB/c cell lysate provided equal protection to the BALB/c islet allografts in B6 recipients compared with infusions of 1 × 108 ECDI-fixed BALB/c SPs (p = 0.72). Control = untreated B6 recipients. (B) Tolerance efficacy with ECDI-fixed A20 cells. Top: A20 cells express BALB/c MHC class I (H-2Kd) and class II (I-Ad). Bottom: infusions of 5 × 107 ECDI-A20 cells significantly prolonged BALB/c islet allograft survival in B6 recipients. Infusions of untreated A20 cells were not able to provide graft protection.
The main donor antigens contained in the lysate important for the induction of donor tolerance are likely donor MHC class I and class II molecules. Therefore, culture-expanded donor cell lines expressing donor MHC I and II, rather than primary donor cells, may possibly be used as a convenient and unlimited source of donor antigens for ECDI coupling for tolerance induction. To test this hypothesis, we examined the A20 cell line for ECDI coupling and tolerance efficacy in the BALB/c to B6 islet transplant model. The A20 cell line is a BALB/c B-cell lymphoma line that expresses both H-2Kd and I-Ad, the MHC I and II of BALB/c, respectively (Fig. 3B, top panels). A20 cells were grown in tissue culture, harvested, and coupled with ECDI as described in Materials and Methods. A dose of 5 × 107 ECDI-A20 cells was injected into diabetic B6 recipients on day −7 and day +1, and islet transplant was performed on day 0 using BALB/c as donors. As shown in Figure 3B, lower panel, injection of ECDI-A20 cells significantly prolonged BALB/c islet allograft survival compared with controls (p = 0.0017). Unexpectedly, two out of nine recipients experienced late rejection around day 90 (Fig. 3B). None of the recipients that rejected their islet allografts showed any evidence of tumor growth by necropsy (data not shown). Currently, it is unclear if the observed difference between tolerance efficacy of the A20 cells and that of BALB/c splenocytes (Fig. 1) is due to A20 tumor cell-specific factors (such as aberrant interactions between the tumor cells and host phagocytes) or due to a lack of other critical cell populations that are present in the SPs, but not in the A20 cells. Nonetheless, this result is a proof of principle that ex vivo expandable cells expressing donor MHC class I and class II molecules can potentially be used to replace primary donor SPs as an alternative source of donor antigens for ECDI coupling for tolerance induction. This finding will now allow future exploration of numerous clinically feasible donor cell products, such as expanded primary donor B-cells, for the use in an ECDI coupling-based tolerance regimen.
Compatibility of Donor ECDI-SP-Based Tolerance Therapy with Conventional Immunosuppressive Drugs
Initial clinical translation of donor ECDI-SP tolerance therapy in human transplant recipients will likely incorporate concurrent use of standard immunosuppressive medications followed by scheduled weaning as in other experimental tolerance protocols (14,15). We have previously shown that the mammalian target of rapamycin (mTOR) inhibitor rapamycin can synergize with donor ECDI-SPs for tolerance induction in a cardiac transplant model (2). Here we tested if the calcineurin inhibitor FK and the purine synthesis inhibitor MMF are compatible with donor ECDI-SP tolerance therapy. FK (3 mg/kg/day, SC) or MMF (100 mg/kg/day, IP) was given from day −1 to day +8 to diabetic B6 recipients, with or without BALB/c ECDI-SPs (given on day −7 and day +1, IV), with islet transplantation performed at day 0 using BALB/c islets. As shown in Figure 4, a short course of FK or MMF treatment alone had a minimal effect on islet allograft survival with all grafts rejected by day 27. Importantly, however, their administration did not have any detrimental impact on the tolerance efficacy of donor ECDI-SPs, with 100% of the islet allograft protected for >60 days. Therefore, commonly used standard immunosuppressive medications are compatible with donor ECDI-SP-based tolerance therapy and can likely be safely incorporated in clinical trial designs testing this tolerance strategy.

Compatibility of donor ECDI-SP-based tolerance therapy with conventional immunosuppressive drugs. Infusions of ECDI-SPs, when combined with a short course (day −1 to day 8) of FK or MMF, remained effective in providing islet allograft protection in 100% of the recipients for >60 days. Control = untreated B6 recipients.
The Spleen Is Dispensable for Transplant Tolerance Induction by Donor ECDI-SPs
Our previous studies have shown that the injected donor ECDI-SPs quickly home to the recipient spleen where they are internalized by recipient APCs including dendritic cells (DCs), macrophages, and B-cells (12). These findings suggest that the spleen might be an important location where the ECDI-SPs exert their tolerogenic effects. To determine whether the spleen is indispensible for tolerance induction by donor ECDI-SPs, we tested the efficacy of ECDI-SPs in splenectomized recipients. The spleen was removed in recipients on day −8, and ECDI-SPs were infused on day −7 and day +1 with day 0 being the day of donor islet transplantation. To our great surprise, splenectomized recipients were equally tolerized by infusions of donor ECDI-SPs and exhibited permanent protection of the transplanted islet allografts (Fig. 5A), suggesting that other anatomical location(s) may act as the site for donor ECDI-SPs to home to and exert their tolerogenic effects in the absence of the spleen.
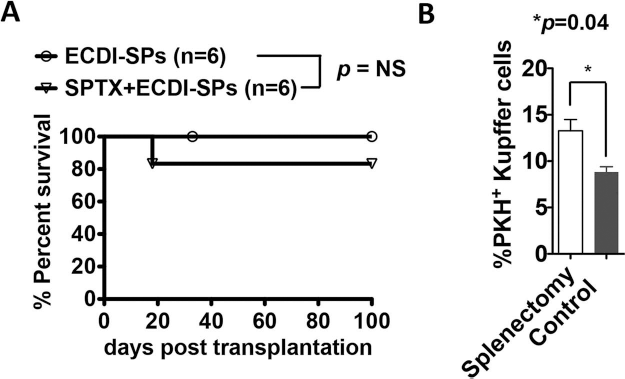
The spleen is dispensable for transplant tolerance induction by donor ECDI-SPs. (A) Islet allografts in splenectomized recipients were equally protected by donor ECDI-SPs as those in nonsplenectomized recipients (p = 0.32). (B) Enhanced uptake of the injected ECDI-SPs by the recipient liver Kupffer cells (IAb+F4/80highCD11blow) in splenectomized recipients compared with that in nonsplenectomized (Control) recipients. PKH-67-labeled donor ECDI-SPs were injected into splenectomized or control nonsplenectomized mice, and 18 h later, the livers were harvested and analyzed. The percentages of PKH-67+ IAb+F4/80highCD11blow cells among all IAb+F4/80highCD11blow cells are shown. Results are the average from three independent experiments of two mice in each group for each experiment.
We have previously noted that second to the spleen, the liver is the next organ to which a large number of the injected donor ECDI-SPs home (12). We hypothesize that in the absence of the spleen (as in splenectomized recipients), the liver becomes the major site where donor ECDI-SPs home to and exert their tolerogenic effects. To examine the cellular distribution of the injected donor ECDI-SPs in splenectomized recipients, we labeled the donor ECDI-SPs with a membrane fluorophore PKH-67 prior to injection, and the distribution of PKH-67+ cells in the liver 18 h postinjection was investigated. As shown in Figure 5B, at 18 h postinjection, a fraction of the liver Kupffer cells (IAb+F4/80highCD11blow cells) were seen to have become PKH-67+, indicating that they had internalized the injected ECDI-SPs. This internalization was significantly increased in splenectomized hosts compared with that in nonsplenectomized hosts. Other anatomical locations including peripheral lymph nodes, the bone marrow, and the lungs did not show significant retention of PKH-67+ fragments (data not shown). We conclude that the spleen is not obligatory for tolerance induction by donor ECDI-SPs infusions. Kupffer cells in the liver are a main cell population capable of internalizing the injected donor ECDI-SPs, particularly in splenectomized recipients, and may consequently play an important role in mediating tolerance induced by donor ECDI-SP infusions in such recipients.
Tolerance Induction by Donor ECDI-SPs Does Not Require CD47 Expression on the Donor SPs
In numerous experimental models of transplantation, donor-specific transfusion (DST) combined with costimulation blockade has been shown to effectively induce donor-specific tolerance (5,6). DST consists of infusion of unmodified donor leukocytes. Recent studies have shown that CD47 expression on the infused donor leukocytes is critically important for tolerance induced by DST, presumably by interacting with the inhibitory receptor signal regulatory protein a (SIRPa) on recipient APCs, thereby modifying their maturation and activation (29). Since CD47 is species specific, incompatibility between donor CD47 and recipient SIRPa impairs tolerance efficacy of DST in xenogeneic transplant tolerance induction, mandating transgenic expression of recipient-species CD47 on the xenogeneic donor leukocytes. To test whether CD47 expression on donor leukocytes is also important for tolerance induction by donor ECDI-SPs, we used donor SPs from CD47-/-mice for ECDI coupling. Because CD47-/- mice are only available on the B6 background, we used a B6 to BALB/c islet transplant model to test the efficacy of CD47-/-donor ECDI-SPs. CD47-/- donor ECDI-SPs were infused on day −7 and day +1 to diabetic BALB/c mice, and B6 islets were transplanted in these mice on day 0. As shown in Figure 6, CD47-/- donor ECDI-SPs were equally efficacious in promoting long-term allogeneic islet graft survival, indicating that CD47 expression on donor SPs is not necessary for their tolerogenic effects. This finding points to a fundamental difference between the tolerance mechanism by ECDI-SPs and that by DST and widens the potential applicability of ECDI-SPs to xenogeneic tolerance induction in humans without the need for engineering human CD47 into donor animals (30).
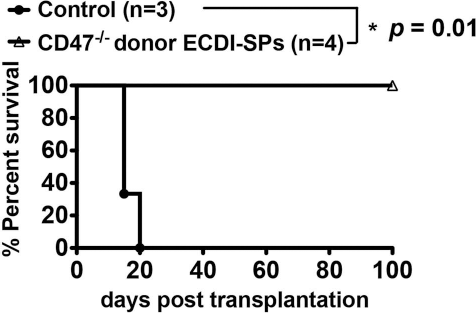
Tolerance induction by donor ECDI-SPs does not require the CD47 expression on the donor SPs. Infusions of CD47-/-donor (B6) ECDI-SPs provided long-term (>100 days) B6 islet allograft survival in 100% of the BALB/c recipients. Control = untreated BALB/c recipients.
Discussion
In this study, we examined the effect of several critical parameters of infusions of donor leukocytes chemically cross-linked with ECDI on the efficacy of transplant tolerance. We show that transplant tolerance can be achieved with a much smaller number of donor cells than initially established and can additionally be achieved with several alternative sources of donor antigens. It is not dependent on the presence of the recipient spleen and is compatible with concurrent usage of common immunosuppressive drugs. Furthermore, distinct from DST, tolerance induction by ECDI-SPs is not dependent on donor cell expression of the CD47 molecule. Tolerance efficacy, however, can be compromised if donor cells undergo a freeze/thaw process, likely due to an increase in necrotic cells induced via this process.
Our findings that expandable cell lines expressing donor MHC antigens, and furthermore cell-free soluble donor antigens, can be used as a substitute for fresh donor cells for ECDI coupling significantly enhance the clinical applicability of this transplant tolerance approach. These not only eliminate the logistic and cost constraints of processing fresh donor cells for each injection of the tolerance therapy but also reduce the variability and burden of quality control of the tolerance products, particularly in situations where repetitive infusions of tolerance products might be necessary. Conceivably, donor B-cells can be expanded ex vivo (25,27). Strategies for large-scale ex vivo B-cell expansion have been experimented using both human and mouse B-cells and can achieve up to 50,000-fold of expansion (10,21). B-cells express both donor MHC class I and class II molecules, and such expressions can be further augmented during the expansion to meet the needs for dose requirement of donor antigen delivery. Therefore, B-cells could be an ideal source of donor cells for the ECDI coupling reaction. Furthermore, the notion of using a cell-free system for tolerogenic donor antigen deliveries can possibly be extended to the use of synthetic bioengineered microparticles for tolerance induction as recently demonstrated in tolerance to autoantigens (7). In this regard, soluble donor antigens can be attached to synthetic microparticles that are then injected into recipients. Theoretical advantages of using synthetic microparticles include the ability of engineering specific molecules, such as apoptotic or inhibitory signals, and/or immunoregulatory cytokines that would further enhance their interaction with the host phagocytic machinery in ways that are most conductive for tolerance induction.
An interesting and somewhat surprising finding from our study using soluble donor lysates coupled to recipient SPs for tolerance induction is that tolerizing the indirect allorecognition pathway appears by itself quite effective (~70%) at providing permanent graft protection in our allogeneic islet transplant model (Fig. 3A). We have previously shown that infusions of ECDI-coupled donor cells target both the direct and the indirect pathways, leading to anergy and deletion of CD4+ T-cells with direct and indirect specificities, respectively (12). The lack of targeting the direct pathway by the donor lysate-coupled recipient SPs might contribute to the incomplete (~70%) graft protection observed (Fig. 3A). It is possible that the indirect pathway is the predominant mechanism of rejection in the islet transplant models we have studied, including allogeneic and xenogeneic islet transplantation. In models in which the direct pathway plays a more important role in rejection (such as due to a larger number of passenger leukocytes carried by the transplanted grafts), this approach of tolerizing the indirect pathway alone by using donor lysates coupled to cellular or cell-free carriers may be less effective and consequently may need to be combined with other approaches to more potently target the direct pathways. Such approaches could include the use of a short course of tolerance-compatible immunosuppression such as those tested in this study, and/or blocking antibodies targeting costimulatory pathways.
Another form of donor-negative vaccination is DST-mediated transplant tolerance (1,8,28). Interestingly, this form of donor negative vaccination also targets recipient DCs to mediate tolerance induction and is exquisitely dependent on the expression of CD47 by the donor cells in a dose-dependent fashion (29,32). DST with CD47-/-donor cells fails to induce transplant tolerance as it leads to the activation of recipient DCs (upregulation of CD86 and MHC class II molecules), whereas DST with CD47+/+ donor cells keep recipient DCs in an immature state, likely through inhibitory signaling via SIRPa (29). This is in sharp contrast to our finding using ECDI-SPs, wherein the tolerance efficacy is completely independent of CD47 expression by the donor cells (Fig. 6). ECDI-SPs also target recipient DCs and upregulate PD-L1 and PD-L2 on the DCs in addition to maintaining a low level of positive costimulatory molecules (12), but mostly likely do so by employing pathways other than CD47-SIRPa. In conjunction with our finding that a favorable apoptotic-to-necrotic cell ratio is necessary for its tolerance efficacy (Fig. 2), we postulate that by progressing to apoptosis rather than necrosis, ECDI-SPs display a set of proper “find-me” and “eat-me” signals that interact with respective receptors on host phagocytes leading to an anti-inflammatory response (20,31). Candidate signals include sphingosine 1-phosphate, fractalkine CX3CL1, lipid lysophosphatidylcholine, and phosphatidylserine, and candidate receptors include scavenger receptors, TIM-4, CD68, and MARCO, among others (7,23). On the contrary, necrotic cells express signals resembling endogenous toll-like receptor ligands, such as high-mobility group box 1 protein, heat-shock proteins, and S100 proteins, leading to the activation and maturation of the recruited host phagocytes (22). This hypothesis points to the obligatory role of apoptosis in the tolerance efficacy of ECDI-SPs, therefore underlining the importance of product specification of ECDI-SP preparations to ensure a high apoptosis to necrosis ratio.
We have previously shown that the injected ECDI-SPs rapidly concentrate in the spleen of the host and interact with several types of phagocytes functioning to silence donor-specific T-cells (12). Therefore, it was surprising that tolerance efficacy was not at all compromised in the absence of the spleen (Fig. 5A). However, in our earlier studies, we also observed that while the spleen was where the predominant ECDI-SPs were captured, a smaller yet significant portion of the ECDI-SPs were also captured in the liver (12). Here we show that this retention in the liver was accentuated when the spleen was removed (Fig. 5B). The liver has long been considered to be a tolerogenic organ due primarily to its continuous exposure to microbial and dietary antigens, hence the need for a robust mechanism for ensuring immune tolerance (9). In addition to the Kupffer cells demonstrated here, other liver nonparenchymal cells such as hepatic DCs, hepatic stellate cells, and liver sinusoidal endothelial cells, as well as liver parenchymal cells, have all been shown to function as APCs (3,4,26). However, the nature of such antigen presentation and its effect on the behavior of the interacting T-cells might be different from those of classical splenic DCs (4,19). Translating to our findings here, it could mean that the mechanisms for tolerance induction by ECDI-SPs in the presence of liver APCs versus spleen APCs are different; consequently the efficiency, stability, and durability of such tolerance may also be different. These aspects warrant future studies in the splenectomized model to thoroughly interrogate the characteristics of tolerance and the behavior of donor-specific T-cells using defined tools.
Meticulous attention has been paid to the design of immunosuppressive regimens for early human trials of tolerance protocols for solid organ transplantation to ensure compatibility between the agents used and known mechanisms of tolerance related to the individual protocols (11, 14,15,24). While some data are available regarding the general pro- or antitolerogenic features of these drugs, their effects on a specific tolerance approach such as ECDI-SPs cannot be easily extrapolated from such generalization. Our data demonstrating the compatibility of a short course of common immunosuppressive drugs FK and MMF with tolerance induction by ECDI-SPs therefore provides the critically important information for the future design of preclinical and clinical studies testing this tolerance approach.
In summary, while the tolerance outcome by the donor ECDI-SP strategy in preclinical nonhuman primate tolerance studies will be difficult to predict, this report provides important and timely information on several critical parameters of this regimen that allow for both effective quality control of the tolerance products as well as for enhancement of efficacy, versatility, and feasibility necessary for the future design of clinical trials employing this novel strategy.
Footnotes
Acknowledgments
We wish to acknowledge the Northwestern University Interdepartmental ImmunoBiology Flow Cytometry Core Facility and the Mouse Histology and Phenotyping Laboratory for their support of this work. This work was supported by grants from the National Institutes of Health Directors New Innovator Award DP2 DK083099 (X.L., S.W., J.B.), NIH U01-AI102463 (B.J.H., X.L., S.D.M.) and the Juvenile Diabetes Research Foundation Postdoctoral Fellowship Grant 3-2010-447 (T.K.). Contributions: S.W., X.Z., T.K., B.J.H., S.D.M., X.L. designed the research; S.W., X.Z., L.Z., J.B. performed the experiments; S.W., X.Z., L.Z., J.B., X.L. analyzed the data; S.W., X.L. wrote the manuscript; B.J.H., S.D.M. edited the manuscript. The authors declare no conflicts of interests.