
Abstract
Cell therapy could potentially meet the need for pancreas and islet transplantations in diabetes mellitus that far exceeds the number of available donors. Bone marrow stromal cells are widely used in clinical trials mainly for their immunomodulatory effects with a record of safety. However, less focus has been paid to developing these cells for insulin secretion by transfection. Although murine models of diabetes have been extensively used in gene and cell therapy research, few studies have shown efficacy in large preclinical animal models. Here we report optimized conditions for ex vivo expansion and characterization of porcine bone marrow stromal cells and their permissive expression of a transfected insulin gene. Our data show that these cells resemble human bone marrow stromal cells in surface antigen expression, are homogeneous, and can be reproducibly isolated from outbred Yorkshire–Landrace pigs. Porcine bone marrow stromal cells were efficiently expanded in vitro to >1010 cells from 20 ml of bone marrow and remained karyotypically normal during expansion. These cells were electroporated with an insulin expression plasmid vector with high efficiency and viability, and secreted human insulin and C-peptide indicating appropriate processing of proinsulin. We showed that autologous insulin-secreting bone marrow stromal cells implanted and engrafted in the liver of a streptozotocin-diabetic pig that modeled type 1 diabetes resulted in partial, but significant, improvement in hyperglycemia that could not be ascribed to regeneration of endogenous β-cells. Glucose-stimulated insulin secretion in vivo from implanted cells in the treated pig was documented by a rise in serum human C-peptide levels during intravenous glucose tolerance tests. Compared to a sham-treated control pig, this resulted in significantly reduced fasting hyperglycemia, a slower rise in serum fructosamine, and prevented weight loss. Taken together, this study suggests that bone marrow stromal cells merit further development as autologous cell therapy for diabetes.
Keywords
Introduction
Biomedical research to develop new treatments is usually initiated in small rodent models of disease. However, methods that are efficacious in inbred strains of laboratory rodents may fail when applied to human subjects because of interspecies biological differences viz., of the immune system, metabolism, anatomy, body size, and natural life span. Limitations of small animal models in translational research are strikingly illustrated by the fact that preclinical studies in large animals were essential and critical in the eventual development and acceptance of transplantation medicine (10). Experiments in dogs, pigs, and nonhuman primates have served importantly to demonstrate the feasibility of transplanting kidney (11), liver (12, 51), heart (17), bone marrow (22, 42), lung (47), small intestine (38), pancreas, and islets (1, 2, 36, 39, 40). Although canine models were used frequently in the past, there are reservations about using companion animals for research, and the use of nonhuman primates for research is restricted, costly, and contentious because of their close evolutionary relationship to humans (46). Other considerations also favor pigs over dogs and nonhuman primates as a large animal model for preclinical studies. Like humans, pigs are outbred, readily procured, and generally healthy. Porcine and human anatomy, body weight, and physiology are highly similar. Moreover, technology now exists to generate and clone transgenic pigs (32, 37).
The success of organ and tissue transplantation has since led to a global demand for replacement of failing organs that far outstrips the supply of donors. Cell therapy could potentially meet this demand for a wide range of disorders such as diabetes, Parkinson's disease, and repair of tissue injury. Different cell types are being investigated for various applications, that is, embryonic stem cells, induced pluripotent stem cells, adult tissue stem cells, and somatic cells. Bone marrow mesenchymal stromal cells (BMMSCs) are the most frequently used cell type, and there are 181 clinical trials listed as using BMMSCs on the US National Institutes of Health registry of clinical trials (ClinicalTrials.gov; accessed on October 28, 2013). In general, these trials attempt to exploit the immunomodulatory, anti-inflammatory, and homing properties of BMMSCs (16, 23, 29, 35) to prevent or suppress allograft rejection (5, 18) and to repair damaged tissues (7, 44, 54, 58).
In contrast to their intrinsic immunological properties, the capacity of BMMSCs to serve as potential cellular carriers of secreted transgenic proteins of therapeutic interest has received rather less attention. Such applications are likely to be feasible as BMMSCs have the capacity to secrete a range of cytokines and proteoglycans (28, 61). Depending on the disorder to be treated, such an application may require a large number of genetically modified BMMSCs. In this regard, substantial proliferation of these cells in vitro is a distinct advantage. Several groups have reported expansion in vitro of primary BMMSCs obtained from a single marrow aspirate to at least several hundred million cells (13, 21, 50, 55, 56).
In previous work, we have shown that primary porcine BMMSCs (pBMMSCs), transfected with an insulin expression plasmid by electroporation, secreted transgenic insulin and improved hyperglycemia and glucose tolerance when implanted in diabetic mice (15). To extend this work toward autologous cell therapy of diabetic pigs, a clinically relevant large animal model, we here describe optimized conditions for ex vivo expansion of pBMMSCs obtained from a single aspirate of bone marrow and electroporation. The expanded and electroporated cell population had a homogeneous immunophenotype and stable karyotype. We established a porcine model of type 1 diabetes by permanently ablating endogenous β-cells with streptozotocin (STZ) and documented the absence of β-cell regeneration. We report the insulin-secreting capacity of expanded pBMMSCs in vitro and demonstrate proof of concept of transgenic insulin secretion in vivo by implanting plasmid-transfected autologous BMMSCs in a diabetic pig.
Materials and Methods
Animals
All animal handling procedures and animal husbandry were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility. The experimental protocol was approved by the SingHealth Institutional Animal Care and Use Committee. Male and female Yorkshire–Landrace pigs (about 3 months of age; body weight approximately 30 kg) were fed standard chow twice daily. Pigs were supplied by the Singapore Experimental Medicine Centre, Singapore.
Cell Culture Surface Optimization
Five different plastic surfaces were evaluated for highest adherence of pBMMSCs: tissue culture-treated polystyrene (#430641; Corning, Corning, NY, USA), proprietary CellBIND® surface (#3290; Corning), a positively charged surface (BD PureCoat™ amine #354726; BD Biosciences, San Jose, CA, USA), a polystyrene surface treated to reduce hydrophobicity (Nunclon™ Delta #156472; Nunc A/S, Roskilde, Denmark), and 0.1% porcine gelatin-coated surface. The latter was prepared by coating each 75-cm2 flask (BD Falcon™, #353136; BD Biosciences) with 15 ml of sterile 0.1% porcine gelatin solution (#G1393; Sigma-Aldrich, St. Louis, MO, USA) for 30 min at 37°C, after which the remaining gelatin solution was removed. Gelatincoated flasks were left to dry at room temperature and stored at room temperature until use.
Porcine BMMSC Isolation and Culture
Twenty milliliters of bone marrow was aspirated from the tibia of a female pig under general anesthesia with a sterile heparinized (0.5 ml heparin solution, 5,000 IU/ml; Hanlim Medical, Seoul, Korea) syringe (#SS-05S; Terumo, Somerset, NJ, USA) and 13-gauge Jamshidi needle (#DJ2013X; CareFusion, San Diego, CA, USA). Anesthesia was induced and maintained with 5% isoflurane (Aerrane; Baxter Healthcare Corporation, Deerfield, IL, USA) inhalation after sedation with intramuscular ketamine (18 mg/kg body weight; Ceva Animal Health, Glenorie, NSW, Australia) and atropine (0.05 mg/kg body weight; Pfizer, Bentley, WA, Australia). Anesthesia was maintained with 2% isoflurane (Abbott Laboratories Limited, Queensborough, Kent, UK) inhalation. Twenty milliliters of whole blood was withdrawn to obtain autologous serum. Bone marrow aspirate was diluted with an equal volume of culture medium. The culture medium was a 3:1 (v/v) mixture of low-glucose Dulbecco's modified Eagle medium (#31600-091; Invitrogen, Carlsbad, CA, USA) and StemPro® (#A10332-01; Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (#SV30160.03; HyClone, Logan, UT, USA), 10,000 units penicillin, and 100 mg streptomycin per liter (#15140122; Life Technologies). Bone marrow in culture medium was filtered through a 400-μm cell strainer (BD Falcon™, R352340; BD Biosciences). The concentration of mononucleated cells (MNCs) was determined after lysing red blood cells (RBCs). This was done by diluting a small aliquot of filtered bone marrow aspirate 20-fold with lysis buffer [0.154 M NH4Cl (#A9434), 0.01 M KHCO3 (#237205), 0.127 mM EDTA (#E6758), all from Sigma-Aldrich] and intermittent vortexing at room temperature for 5 min before counting MNCs in a Neubauer hemocytometer chamber (#442/12; Glaswarenfabrik Karl Hecht GmbH & Co. KG, Sondheim von derRhon, Germany).
To evaluate five different culture surfaces, 4 ml of filtered bone marrow aspirate mixed with 6 ml of culture medium was seeded in each 75-cm2 flask. Nonadherent cells were removed after 72 h, adherent cells were washed with phosphate-buffered saline (PBS) [137 mM NaCl (#1.06404.5000), 2.7 mM KCl (#1.04936.1000), 8.1 mM Na2HPO4 (#1.06586.2500), 1.5 mM KH2PO4 (#1.04873.1000), all from Merck KGaA, Darmstadt, Germany], and fresh culture medium was added. Cell counts were performed 7 days after the initial seeding.
For large-scale cell expansion, bone marrow aspirates were seeded at a density of 3–5 × 105 MNCs/cm2 in culture medium in 175-cm2 Nunclon™ Delta treated flasks (#159910; Nunc A/S). Nonadherent cells were removed after 72 h. Adherent cells were washed once with PBS before adding fresh medium. Thereafter, culture medium was changed every third day. Cells were trypsinized at 80% confluence with TrypLE™ Express (#12604021; Life Technologies). Thereafter, pBMMSCs were either further expanded or cryopreserved in culture medium containing 7% dimethyl sulfoxide (#D2650; Sigma-Aldrich) and 20% fetal bovine serum. Multilayer flasks (Nunc™ TripleFlasks, #132913; Nunc A/S) were used during later stages of cell expansion for work efficiency.
Immunophenotyping
pBMMSCs of six pigs (three males and three females) were immunophenotyped for surface antigen expression (30). Cells from early passage, late passage, and after electroporation were trypsinized at 80% confluence, washed with PBS, and counted. Cell suspensions (5 × 105 cells in 500 μl) were filtered through a 40-μm mesh strainer (#03-41/31; Sefar AG, Heiden, Switzerland) into individual tubes containing the following antibodies and incubated at 4°C in the dark for 30 min, with gentle flicking every 10 min. Antibodies used were CD105–PE (20 μl/106 cells; #ab53321; Abcam, Cambridge, UK), IgG2a–PE (20 μl/106 cells; #ab91363; Abcam), CD90–PE (5 μl/106 cells; #555596; BD Pharmingen, San Jose, CA, USA), IgG1, κ–PE (20 μl/106 cells; #555749; BD Pharmingen), CD29–PE (20 μl/106 cells; #ab64629; Abcam), IgG1–PE (10 μl/106 cells; #ab81200; Abcam), CD11b–APC (20 μl/106 cells; #301310; BioLegend, San Diego, CA, USA), IgG1, κ–APC (5 μl/106 cells; #400120; BioLegend), HLA DR + HLA DP (HL-38)–FITC (20 μl/106 cells; #ab18232; Abcam), IgG2a–FITC (10 μl/106 cells; #MCA929F; AbDF Serotec, Kidlington, UK), CD14–FITC (10 μl/106 cells; #MCA1568F; AbD Serotec), CD79α–FITC (10 μl/106 cells; #MCA2538F; AbD Serotec), IgG1–FITC (10 μl/106 cells; #MCA928F; AbD Serotec), and CD45–FITC (10 μl/106 cells; #MCA1222F; AbD Serotec).
Cells were washed with PBS, resuspended in 500 μl of cold staining buffer (2 mM EDTA, 0.5% FBS in PBS), and analyzed in a BD FACSCalibur™ flow cytometer (BD Biosciences). Data analysis was performed using FlowJo 7.0 (TreeStar, Ashland, OR, USA).
Karyotyping
pBMMSCs of three pigs (two males and one female), cryopreserved at various passages, were reestablished in culture for a week and sent to the Genomic Institute of Singapore for G-banded karyotyping.
Small-Scale Cell Electroporation
pBMMSCs resuspended in 100 μl of P1 solution (Amaxa #V4XP-1024; Lonza, Basel, Switzerland) with plasmid DNA (2.5 μg/106 cells) were electroporated using the 4D-Nucleofector™ System (FP100 program; Lonza). One hundred microliters of culture medium was added immediately after electroporation to the cuvette, and cells were incubated at 37°C for 10 min.
Plasmid Preparation
pTopo3EGR1chINS (15), pcDNA™3.1 (Life Technologies), and pEGFP-C1 (Clonetech, Mountain View, CA, USA) DNA were prepared using EndoFree Plasmid Giga Kit (#12391; Qiagen, Hilden, Germany).
Optimizing Cell Number for Electroporation
pBMMSCs (5 × 106, 10 × 106, and 20 × 106) were electroporated with pEGFP-C1. The number of viable pBMMSCs was counted after 10 min. Transfection efficiency was determined 24 h after electroporation by flow cytometry analysis of GFP-positive cells (BD FACSCalibur™ flow cytometer and CellQuest™ Pro software; BD Biosciences).
Large-Scale Cell Electroporation for In Vivo Implantation
Large-scale electroporation of pBMMSCs for in vivo implantation was performed as follows. Batches of 1 × 108 pBMMSCs were pelleted by centrifugation at 200 × g for 5 min in 50-ml polypropylene tubes (#227261; Greiner Bio-One, Frickenhausen, Germany) and then gently dispersed by manually tapping the tube. The electroporation medium was prepared separately by mixing 450 μl of P1 solution and 50 μl plasmid solution containing 250 μg pTopo3EGR1chINS (15) in a homopolymer microfuge tube (#MCT-150-C; Axygen Inc., Union City, CA, USA). The complete electroporation medium thus prepared (500 μl) was added to the dispersed cell pellet and gently mixed. Electroporation was performed using the Lonza 4D-Nucleofector™ System (Lonza Cologne GmbH, Koln, Germany), proprietary cuvettes and P1 solution provided in the P1 Primary Cell 4D-Nucleofector™ X Kit L (#V4XP-1024; Lonza Limited, Basel, Switzerland). Each cuvette was filled with 140 μl of the cell suspension in electroporation medium and electroporated using the FP100 program (Lonza Limited). About 100–120 individual electroporations were performed to transfect a total of 2 × 109 pBMMSCs. Electroporated pBMMSCs were mixed with autologous serum for immediate implantation in vivo.
A small aliquot of pBMMSCs electroporated with pTopo3EGR1chINS was set aside and replated in insulin-free culture medium that was changed after 24 h. Conditioned medium 48 h after electroporation was assayed for human insulin and human C-peptide (HCP) to confirm that the implanted cells were capable of secreting insulin.
STZ-Induced Diabetes
Pigs (one male and one female) were fed ammonium chloride (1.5 g/kg body weight in 500 g moistened chow; VWR International, Radnor, PA, USA) 16 h before a bolus ear vein injection, over 4 min, of STZ (120 mg/kg body weight; #ALX-380-010-G001; Enzo Life Sciences, Farmingdale, NY, USA) followed immediately by 30 ml of isotonic saline (B. Braun Medical Industries, Penang, Malaysia). STZ was dissolved in fresh citrate buffer, pH 4.5, on the day of injection. Citrate buffer was prepared by mixing 27.85 ml of 0.1 M disodium citrate solution [2.101 g monohydrated citric acid (#1.00244.0500) dissolved in 20 ml of 1 M NaOH (#106498) and brought to 100 ml with water] with 22.15 ml of 0.1 M HCl (#1003172500). All citrate buffer reagents were from Merck (KGaA, Darmstadt, Germany). Once daily subcutaneous injections of insulin glargine (Lantus®; Sanofi Aventis, Paris, France) were administered before surgery to moderate severe hyperglycemia. Insulin injections were discontinued immediately after autologous pBMMSC implantation.
In Vivo Cell Implantation
Under general anesthesia, electroporated pBMMSCs were injected directly into multiple sites in the liver. A diabetic female pig was implanted with 2.1 × 109 autologous pBMMSCs electroporated with pTopo3EGR1chINS. The total volume of injected cells injected into six liver sites was 21.5 ml. Five sites in the right medial lobe were each injected with 4 ml of electroporated pBMMSCs. One site in the caudate lobe was injected with 1.5 ml. A diabetic male pig that served as the untreated control was implanted in three sites in the right medial lobe (3 ml each) with 0.9 × 109 autologous pBMMSCs electroporated with pcDNA3.1.
Pancreas Biopsies and Immunohistochemistry
Serial biopsies of the pancreas were performed laparoscopically (Karl Storz, Tuttlingen, Germany) under general anesthesia in two male pigs before, 1-3 weeks, and 3–4 months after STZ-induced diabetes. An intravenous glucose tolerance test (IVGTT) was performed at the time of each biopsy to determine blood glucose and serum porcine C-peptide (PCP) responses.
Slides were prepared of 4-μm sections of formalin-fixed paraffin-embedded pancreatic tissues from six blocks obtained at each state, that is, normal nondiabetic, acute diabetes, and established diabetes for each pig. Tissue sections were incubated with mouse monoclonal anti-porcine insulin antibody (sc-8033; Santa Cruz Biotechnology, Dallas, TX, USA), after which a chromogenic reporter generated by the peroxidase-diaminobenzidine reaction stained antibody-bound cells (REAL™ EnVision™ Detection System, #K5007; Dako, Glostrup, Denmark). All sections were counterstained with hematoxylin (#H-3404; Vector Laboratories, Burlingame, CA, USA). Stained tissue sections were scanned at 20× magnification (Scan-Scope CS2; Aperio Technologies, Vista, CA, USA), and quantitative analysis of insulin-positive cells was performed with ImageScope version 10 (Aperio Technologies), using a pixel-counting algorithm. Only brown pigments from the peroxidase-diaminobenzidine reaction were set as positive in the default input parameters. Using the grid option, all pixels in three contiguous 800-μm2 squares in each tissue section were analyzed and counted (3.0× zoom). Data are expressed as the percentage of positive pixels in each section.
Intravenous Glucose Tolerance Test
We performed IVGTTs on 16-h fasted animals, that is, two pigs (one male and one female) that were implanted with autologous pBMMSCs (“In Vivo Cell Implantation™ above). An internal jugular vein was cannulated (7-French catheter; Arrow International, Inc., Reading, PA, USA) under general anesthesia. To establish the baseline, an isotonic saline bolus (equal to the volume of glucose solution) was injected over 4 min, and blood was drawn 10, 20, and 30 min later for blood glucose, plasma HCP, and PCP assays, as indicated. This was followed by a bolus glucose injection (1 g/kg, 50% w/v solution; B. Braun Medical Industries, Penang, Malaysia) over 4 min followed by 30 ml of saline injection to flush the line. Two milliliters of blood was drawn 1, 3, 5, 10, 20, 30, 45, 60, 75, and 90 min after the glucose bolus for the same assays.
Blood glucose concentrations during IVGTT and daily fasting blood glucose (FBG) concentrations were measured using a glucometer and test strips (ACCU-CHEK® Advantage; Roche Diagnostics GmbH, Mannheim, Germany).
Insulin and C-Peptide Immunoassays
Human insulin and HCP were measured in conditioned media of electroporated pBMMSCs using ultrasensitive human insulin radioimmunoassay (#HI-11K; Millipore, Billerica, MA, USA) and HCP radioimmunoassay (#HCP-20K; Millipore) kits. Results are expressed as total 24-h secretion per 106 cells calculated by multiplying insulin or HCP concentration by the volume of conditioned medium and normalized to 106 cells.
Porcine sera for C-peptide assays were prepared from whole blood samples collected in aprotinin (#616371; final concentration 520 KIU/ml; Millipore) during IVGTT. Serum HCP was measured using a radioimmunoassay that did not cross-react with PCP and human insulin and had <4% cross-reactivity with human proinsulin. Serum PCP was measured using a solid phase two-site enzyme immunoassay (#10-1256-01; Mercodia, Uppsala, Sweden) that had the following cross-reactivities: <0.003% human C-peptide, <0.05% porcine insulin; <0.01% human proinsulin, and <0.02% porcine proinsulin.
Serum samples whose counts on the HCP radioimmunoassay were between zero and the lowest HCP calibrator standard (0.156 ng/ml) were assigned half the concentration of the lowest standard calibrator, that is, 0.078 ng/ml for area under the curve (AUC) calculations.
Statistical Analysis
Data are mean ± standard deviation of duplicate values. Differences were considered significant if p < 0.05 by Student's t-test. The Bonferroni correction was applied for multiple comparisons. GraphPad Prism was used for statistical analyses (GraphPad Software Incorporated, San Diego, CA, USA).
Results
Evaluation of Plastic Surfaces and Red Blood Cell Lysis for Highest Attachment of pBMMSC
Mesenchymal stromal cells are conventionally obtained by adherence to plastic. However, the current range of different coatings and modifications of virgin polystyrene made it necessary to identify the specific surface that favored highest attachment of pBMMSCs. Of the five different surfaces evaluated, the number of pBMMSCs obtained by plating bone marrow on the proprietary Nunclon™ Delta surface was significantly higher (p < 0.005) than on all other surfaces tested (Fig. 1A). Thus, subsequent pBMMSC cultures were performed on this surface only.

Optimizing initial pBMMSC culture conditions. (A) Attachment of pBMMSCs from a female pig to different surfaces: (a) Corning standard tissue culture-treated surface; (b) BD PureCoat™ amine; (c) Corning CellBIND®; (d) porcine gelatin-coated surface; and (e) Nunclon™ Delta. Data are mean ± SD of duplicates. *Significantly different from surface e (Nunclon™ Delta) (Bonferroni-corrected p ≤ 0.005 for all comparisons). (B) Red blood cell lysis of bone marrow aspirated from a female pig before plating on Nunclon™ Delta did not enhance attachmentand establishmentof pBMMSC cultures (p = 0.32). Data are mean ± SD of duplicates.
To determine if RBC lysis before culture would increase the yield of pBMMSCs, bone marrow aspirate was divided into two aliquots of 1.6 ml each (7.8 × 106 MNC/ml). One aliquot was treated with RBC lysis buffer twice, and cells were washed with PBS before plating. The other aliquot was directly plated without RBC lysis. Culture medium was changed after 3 days, and adherent cells were washed with PBS. The numbers of pBMMSCs obtained from both groups on day 8 were not statistically different (p = 0.32) (Fig. 1B), showing that RBC lysis step did not enhance the isolation of pBMMSCs.
In Vitro Expandability of pBMMSC Cultures
The average number of MNCs in bone marrow aspirates was 14.6 × 106 ± 4.5 × 106 (mean ± SD) per ml bone marrow. After removal of nonadherent cells on day 3, the plastic surface was covered with numerous small clusters of pBMMSCs, which rapidly proliferated to dense clusters. As early as the second passage, up to 1–3 × 109 cells were obtained even when not all of the original bone marrow aspirate was expanded. If all the initial pBMMSC clusters had been expanded, the projected final cell number of pBMMSCs after 11–20 days from three different pigs, based on the average fold increase during passaging was 11.8 × 109–37 × 109 pBMMSCs per animal (Table 1).
Expandability of pBMMSCs in Culture

P0 was the stage when newly formed cell clusters from the bone marrow aspirate were first trypsinized.
Final number of cells expanded from part of the initial bone marrow aspirate.
Projected total number of cells if the entire initial bone marrow aspirate had been expanded in culture.
Large-Scale Electroporation
Large-scale electroporation was investigated in order to facilitate implantation of 1–2 × 109 plasmid-transfected cells. Different numbers (5 × 106, 10 × 106, and 20 × 106) of pBMMSCs, resuspended in 100 μl of P1 solution, were electroporated with pEGFP-C1 DNA (9 μg/μl). Cell viability determined 30 min postelectroporation by trypan blue exclusion and transfection efficiency assessed by GFP positivity after 24 h remained high (>85% and >90%, respectively) even for the highest cell density electroporated (2 × 107cells per 100 μl) (Table 2). This cell density was therefore used in subsequent experiments.
Large-Scale Electroporation of pBMMSCs With pEGFP-C1 Plasmid
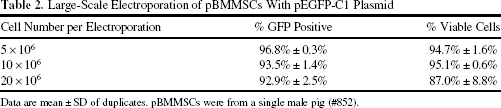
Data are mean ± SD of duplicates. pBMMSCs were from a single male pig (#852).
Immunophenotype and Karyotype of pBMMSC
pBMMSC cultures reestablished from cell stocks cryopreserved at early (P1) and late (P5) passages and after electroporation were immunophenotyped by flow cytometry for positive markers (CD105, CD29, CD90) and negative markers (CD11b, CD45, CD79a, CD14, and HLA DR/DP) of mesenchymal stromal cells (30). Figure 2 shows the pBMMSC immunophenotype of one representative animal displaying the predicted profile of positive and negative markers. Table 3 summarizes the immunophenotype data of pBMMSCs of six pigs. We noted some variation in expression of CD105 and CD45 among pBMMSCs isolated from six pigs. CD105, a positive marker, was expressed in 70–80% of pBMMSCs from three pigs, but in nearly 100% of pBMMSCs from the other two pigs. CD105 positivity of the pBMMSCs of pig #795 (Table 3) actually increased to >96% as it was expanded in culture. CD45, a negative marker, was expressed by 20% of pBMMSCs from two pigs. Overall, the surface marker expression profile of pBMMSCs in all three stages (early passage, late passage, and postelectroporation) did not display marked differences, indicating a homogeneous cell population having a stable immunophenotype.

Expression of pBMMSC surface markers by immnunophenotyping. Representative immunophenotype of one of six pigs (see Table 3). pBMMSCs from a female pig (#850) at passage one, passage five, and postelectroporation were stained with antibodies against the porcine cell surface antigens indicated and analyzed by flow cytometry. pBMMSCs were strongly positive for CD105, CD29, and CD90 and showed minimal or no expression of CD11b, CD45, CD79a, CD14, and HLA DR/DP throughout culture expansion and after electroporation.
Immunophenotypes of pBMMSCs Isolated From Six Pigs
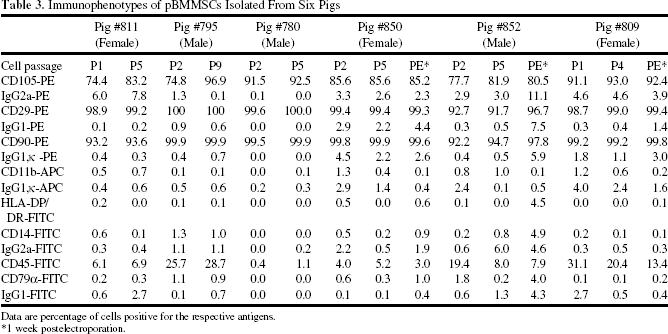
Data are percentage of cells positive for the respective antigens.
1 week postelectroporation.
pBMMSCs from three pigs were karyotyped at early (P1 and P2) and late (P4–P9) passages. For one pig (female), karyotyping was also performed on pBMMSCs that were briefly recultured after electroporation. For each stage of cell expansion, 20 metaphases were randomly analyzed, and all had the normal karyotype of 76 chromosomes. This showed that pBMMSCs were also genomically stable throughout significant cell expansion in vitro and that electroporation did not induce chromosomal abnormalities (Fig. 3).

Karyotyping of pBMMSCs. Karyotypes of pBMMSCs isolated from three pigs (#780 male, #790 male, and #809 female) were consistently normal when examined at early and late passage and postelectroporation.
Nonregeneration of β-cells After Streptozotocin-Induced Diabetes
Serum PCP levels during IVGTT (expressed as AUC20–90 min) were used to assess endogenous β-cell insulin secretion in pigs before and after STZ-induced diabetes. At the dose administered, STZ (120 mg/kg body weight) rapidly and almost completely abolished secretion of endogenous insulin within 1 week, and this remained unchanged for at least 9 weeks when the animals were euthanized. Serum PCP levels were, at most, about 1% of normal levels in established STZ-induced diabetes (Table 4; Fig. 4A–C). This was evidence of irreversible and near-total ablation of endogenous β-cells. To further confirm that endogenous β-cells did not regenerate, serial biopsies of the pancreas were performed in two male pigs. Pancreatic tissue sections stained with anti-porcine insulin antibody showed a 92% reduction of insulin-positive cells in early and established diabetes compared to equivalent sections obtained before STZ administration (i.e., in the nondiabetic state) (Figs. 4D and 5). Both PCP and quantitative immunohistochemical data were evidence that the residual pancreas was incapable of regenerating β-cells under these conditions (Table 5).

Serum PCP and quantitative immunohistochemistry show irreversible ablation of endogenous porcine β-cells. (A, B) Serum PCP responses of pig #862 male and pig #870 male during IVGTT in the nondiabetic, newly diabetic, and established diabetic states showing total absence of PCP response when diabetes was induced by STZ. (C) PCP AUC20–90 min of pig #862 and pig #870 calculated from IVGTT data in the three aforementioned states. (D) Quantitative analysis of cells positively stained with anti-porcine insulin antibody in pancreas tissue sections obtained by biopsy in the three aforementioned states.

Immunohistochemical staining of pancreatic tissue biopsies for endogenous β-cells. Pancreas was biopsied when the pigs (#862 male and #870 male) were normal (nondiabetic), newly diabetic (1 week after STZ injection), and when diabetes was well established (9–12 weeks after STZ). Tissue sections were stained with monoclonal anti-porcine insulin antibody whose binding to β-cells was visualized by a chromogenic reporter generated by the peroxidase-diaminobenzidine reaction. Scale bar: 1,000 μm.
Serum PCP of Four Pigs During IVGTT Performed in the Normal Nondiabetic State, Newly Induced Diabetes, and Established Diabetes

One week after STZ injection for all pigs, except pig #809 (7 weeks after STZ injection).
Nine to 15 weeks after STZ injection.
Summary of Counts of Positive Pixels in Pancreas Sections Stained With Anti-Porcine Insulin Antibody and Serum PCP Levels During IVGTT Expressed as AUC20–90 min of Two Pigs Examined in the Nondiabetic State, in the Newly Diabetic State, and After Diabetes Was Well Established

One week after STZ injection for both pigs.
Nine to 12 weeks after STZ injection.
Insulin Secretion In Vitro by Electroporated pBMMSC
pBMMSCs isolated from five pigs (one male and four female) were electroporated with pTopo3EGR1chINS plasmid DNA. Conditioned media were assayed for human insulin or HCP using the appropriate radioimmunoassays as described. Table 6 shows that pBMMSCs expressed and secreted processed insulin. The variable amounts of insulin or HCP secretion by pBMMSCs of different animals were likely due to differences in transfection efficiency. Nonetheless, we estimated that these levels of insulin secretion could, given a sufficient cell number, result in detectable secretion in vivo.
Twenty-Four Hour Secretion of Insulin and HCP Ex Vivo per 1 ×106 pBMMSCs

Effects of In Vivo Implanted Insulin-Secreting Autologous BMMSCs in a Streptozotocin-Induced Diabetic Pig
Diabetic pig #809 (female) was implanted in the liver with 2.1 × 109 autologous pBMMSCs electroporated with pTopo3EGR1chINS. Control untreated diabetic pig #821 (male) was implanted with 0.9 × 109 autologous pBMMSCs electroporated with a null plasmid, pcDNA3.1. Serum HCP and PCP assays were performed on IVGTT blood samples collected in the nondiabetic state, 1 week after cell implantation and thereafter, at fortnightly intervals. No insulin was administered to pig #809 after implantation. However, untreated diabetic pig #821 received a total of 238 IU of subcutaneous insulin glargine to mitigate severe hyperglycemia over the 80-day period after implantation of autologous cells electroporated with a null plasmid.
FBG of STZ-diabetic pigs was monitored daily before and once weekly after cell implantation. Serial FBG values of treated pig #809 decreased significantly from 21.9 ± 2.4 mM (mean ± SD; n = 11) before cell implantation to 16.5 ± 2.6 mM (n = 8) over the 68-day period after implantation (p = 0.0002) without any injections of exogenous insulin. In contrast, serial FBG values of the sham-treated control pig #821 before (24.0 ± 4.2 mM; n = 10) and after cell implantation (27.7 ± 3.8 mM; n = 7) were not different statistically (p = 0.4586), even though this animal required multiple injections of subcutaneous insulin glargine over the same period to control severe hyperglycemia (Fig. 6). This showed that insulin-secreting pBMMSCs had engrafted in vivo as an autologous implant and had some efficacy in reducing severe hyperglycemia.

Effect of implantation of autologous pBMMSC in vivo on fasting hyperglycemia of diabetic pigs. Upper: Time course of FBG values. Values recorded before STZ-induced diabetes are plotted against days assigned negative numbers (left of the y-axis). STZ was administered on day 1. Cell implantation was performed on day 32 for pig #809 and day 17 for pig #821. Data of pigs #809 and #821 are in blue and red, respectively. Lower: Summary table of mean and standard deviation of FBG values of diabetic pigs #809 and #821 pre- and post-cell implantation. Also shown is the total amount of insulin glargine that was administered to mitigate hyperglycemia of pig #821 after sham treatment.
Serum HCP in multiple samples of pig #809 before implantation consistently fell below the lowest calibrator standard of the radioimmunoassay, but became clearly detectable in all samples after cell implantation. Human insulin secreted by implanted autologous pBMMSCs in this animal was quantified by HCP AUC20–90 min during serial IVGTTs. Table 7 shows that HCP secretion rose by more than 3.5-fold after cell implantation and remained elevated for at least 48 days. Data from the sham control pig #821 showed evidence of a spurious interference with the HCP radioimmunoassay as a false-positive AUC20–90 min was obtained even in the animal's native state (i.e., before any in vivo procedure had been performed). In contrast to pig #809, the spurious serum HCP of this animal actually decreased after cell implantation (Table 7).
Summary of Serum HCP, Serum PCP, and Blood Glucose Responses During IVGTT Expressed as AUC20–90 min of Treated Pig #809 Implanted With 2 × 109 pBMMSC Electroporated With pTopo3EGR1chINS and of Sham-Treated Control Pig #821 Implanted With 9 × 108 Autologous pBMMSC Electroporated With pcDNA3.1

Number of days relative to cell implantation day.
N.D., not done.
Trends in serum fructosamine concentrations and body weights were consistent with some degree of therapeutic efficacy achieved by implanting autologous insulin-secreting pBMMSCs. Serum fructosamine concentration of pig #809 rose from 624 μM on the day of cell implantation when the animal had been rendered diabetic to 681 μM after 49 days (i.e., 9% increase), reflecting a continuing state of hyperglycemia. In contrast, serum fructosamine of pig #821 rose more markedly from 437 μM to 630 μM and 757 μM 28 and 61 days, respectively, after sham treatment, that is, increases of 44% and 73%. Similarly, the degree of body weight loss during the study period was different, that is, the treated pig did not lose as much body weight as its sham control. The body weight of pig #809 69 days after cell implantation was 96% of its body weight on the day of implantation (37 kg vs. 38.5 kg), whereas the body weight of pig #821 70 days after cell implantation was 79% of its body weight on the day of implantation (37 kg vs. 47 kg).
Discussion
The supply of whole pancreata and islets suitable for transplantation in patients with diabetes cannot realistically meet the global demand for such therapy. Several different research approaches are being intensively pursued as attempts to develop cell therapies that could potentially circumvent the short supply of donors and restore near-normal insulin secretion in vivo. An approach widely considered to have great future promise is directed differentiation of embryonic stem cells and induced pluripotent stem cells into functional β-cells or pancreatic islets (20). Direct reprogramming of pancreatic exocrine cells into β-like cells has been reported (64) by the same laboratory that more recently identified a liver-derived factor, betatrophin, that stimulated proliferation of residual β-cells in diabetic animals (63). A recent report has shown differentiation of human BMMSCs into islet-like cell clusters that expressed insulin, C-peptide, and glucagon, which restored euglycemia when transplanted into diabetic mice (25).
A possible disadvantage of cell therapy based on regenerating new β-cells is the known recurrence of underlying autoimmunity that caused β-cell loss in the first instance (8, 26, 53, 59, 62). Our strategy has thus been to develop autologous non-b-cells for restoring insulin production in vivo. In addition to ameliorating hyperglycemia and its associated complications, this strategy, if successful, would not incur the risks of chronic immunosuppression in recipients of allogeneic transplants and eliminate the need for donor organs. In previous work, we showed significant improvement in the metabolic status of diabetic pigs implanted with autologous primary hepatocytes electroporated ex vivo with an insulin expression plasmid (14). The goal of our present work is to investigate the feasibility of using autologous primary BMMSCs as an alternative to hepatocytes because they can be obtained less invasively, are efficiently expanded in culture, and therefore more likely to be acceptable for clinical cell therapy.
BMMSCs are extensively used in regulated clinical trials. One hundred and eighty-one investigational studies of BMMSCs in human subjects are currently registered in the US National Institutes of Health database (clinicaltrials.gov; accessed on October 28, 2013). Of these, six trials are directed at treating type 1 and type 2 diabetes, but none involves transplanting autologous insulin-secreting BMMSCs. Primary mesenchymal stromal cells have a record of safety in clinical trials (6, 33, 60). Several characteristics make them an appealing choice for cell therapies. They are secretory and immunomodulatory cells that also exert paracrine effects, which may facilitate tissue repair and regeneration (31). As potential mediators for restoring insulin secretion in vivo, the facile expansion of autologous BMMSCs ex vivo is a distinct advantage (48). The significance of chromosomal abnormalities found in ex vivo expanded BMMSCs is unsettled (4), although the weight of current opinion is that the observations do not signal a significant clinical risk if primary cells that are not immortalized (i.e., will senesce) are used for therapy (45, 49).
While there are numerous reports of gene and cell therapy of diabetes in small animal models, advancing this large body of work by demonstrating scalability to clinical trials will be more soundly justified by evidence of efficacy in large preclinical animals. The fact that reports of gene and nonislet cell treatment in large diabetic animals are very sparse (9, 14, 43) highlights both the considerable challenges of large animal models and the urgent need to successfully scale-up small animal techniques as a prerequisite for human subject research. There are already preclinical studies of porcine BMMSCs in repair of skin (24), bone (52, 65), articular cartilage (34), and ischemic myocardium (57). Our data show that porcine BMMSCs are similar to human BMMSCs (Fig. 2 and Table 3). Although BMMSCs are operationally isolated as plastic-adherent cells, this empirical method may capture a heterogeneous population of cell types. Our methods reproducibly isolated and expanded homogeneous BMMSC populations in culture from different pigs whose profiles of surface marker expression by immunophenotyping were concordant with defining criteria for mesenchymal stromal cells established by the International Society for Cellular Therapy (19). Our data also showed very substantial proliferative capacity of pBMMSCs in culture, from which more than 1010 cells from 20 ml of marrow aspirate could readily have been generated (Table 1). Moreover, under our conditions of culture expansion, pBMMSCs maintained a normal karyotype through serial passaging and after electroporation, indicating a high degree of genomic stability. These collective features suggest that pBMMSCs are useful in preclinical in vivo trials of cell therapy.
BMMSCs must secrete mature processed insulin in order to serve as clinically useful bioimplants in diabetes. In this study, pBMMSCs were electroporated with pTopo3EGR1chINS, which encodes human proinsulin cDNA modified to enable cleavage of the prohormone by furin, with release of human C-peptide (15). Secretion of both human insulin and C-peptide by electroporated pBMMSCs confirmed that these cells are competent in processing proinsulin (Table 6). Results obtained from implanting autologous insulin-secreting pBMMSCs into a diabetic pig document synthesis, processing of transgenic human proinsulin, and secretion of mature human insulin in vivo. Although normoglycemia was not restored in the treated diabetic pig, our data provide several lines of evidence that a significant degree of metabolic correction was achieved. First, mean FBG concentrations declined significantly in the treated pig (21.9 mM to 16.5 mM) compared to the sham-treated control (Fig. 6). Second, serial IVGTT showed significant glucose-stimulated human insulin secretion, quantified by serum HCP AUC20–90 min, compared to the sham-treated diabetic pig (Table 7). Third, severity of hyperglycemia (measured as blood glucose AUC20–90 min during IVGTT) increased 2.4- and 2.6-fold over the normal nondiabetic state 29 and 61 days, respectively, after sham treatment (Table 7), whereas the same index increased by only 1.8- and 1.3-fold in the treated pig 34 and 48 days, respectively, after cell implantation (Table 7). Fourth, serum fructosamine (a time-integrated measure of hyperglycemia) (3, 27) of the treated pig before and 80 days after implantation of insulin-secreting cells (624 and 611 μM, respectively) was very similar. In contrast, serum fructosamine of the sham-treated pig 84 days after implantation (664 μM) had increased 2.7-fold over the concentration on the day of sham treatment (243 μM). Fifth, weight loss is a feature of severe type 1 diabetes. In this respect, the body weight of the treated pig showed little reduction (4%) at the end of the study compared to the day of cell implantation, whereas the sham-treated pig's body weight over a comparable period was 21% lower. Having documented irreversible destruction of endogenous porcine β-cells by STZ in this study (Tables 4 and 5; Figs. 4 and 5), the foregoing data can reasonably be ascribed to human insulin expressed by autologous pBMMSCs in the treated animal.
A challenge we encountered in scaling-up autologous cell therapy was maintaining high viability of cells during the process of multiple serial electroporations and cell processing after electroporation. Our current workflow took about 6 h to complete, by which time cell viability decreased from >90% to about 50%. This is being addressed in ongoing experiments to shorten the duration of and minimize steps in cell handling (such as centrifugation) to maintain high viability of implanted cells that could result in greater metabolic correction. It is worth noting, however, that the absolute reduction in mean FBG of 5.4 mM achieved in this study is not trivial in the clinical context of diabetes management. For example, a decrease in average blood glucose from 11.8 mM to 7.0 mM is likely to be accompanied by a decrease in glycated hemoglobin, HbA1C, from 9% to 6%–-the latter being a clinical index of good glycemic control (41).
We envisage future developments that combine nonviral gene transfer with techniques for genome site-specific targeted transgene integration, such as zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats/Cas 9 nuclease, could extend the activity of plasmidencoded insulin transgenes, while mitigating the risks of insertional mutagenesis inherent in integrating viral vectors.
In summary, this study describes culture conditions for efficient expansion of primary porcine BMMSCs and shows their similarity to human BMMSCs. Porcine BMMSCs are genomically stable, can be transfected efficiently with plasmid DNA by electroporation, and are capable of synthesizing and secreting human insulin. We show that autologous insulin-secreting porcine BMMSCs engrafted in vivo and ameliorated the metabolic abnormalities of a severely diabetic pig. These results point to the feasibility of further developing autologous cell therapy in a large preclinical animal model of type 1 diabetes.
Footnotes
Acknowledgments
This study was funded by the National Medical Research Council, Singapore (NMRC/1243/2010). We thank Jaichandran S. for critical reading of the manuscript. The authors declare no conflicts of interest.