
Abstract
The polyglutamine (polyQ) diseases are a group of neurodegenerative disorders caused by expanded cytosine– adenine–guanine (CAG) repeats encoding a long polyQ tract in the respective proteins. To date, a total of nine polyQ disorders have been described: six spinocerebellar ataxias (SCA) types 1, 2, 6, 7, 17; Machado–Joseph disease (MJD/SCA3); Huntington's disease (HD); dentatorubral pallidoluysian atrophy (DRPLA); and spinal and bulbar muscular atrophy, X-linked 1 (SMAX1/SBMA). PolyQ diseases are characterized by the pathological expansion of CAG trinucleotide repeat in the translated region of unrelated genes. The translated polyQ is aggregated in the degenerated neurons leading to the dysfunction and degeneration of specific neuronal subpopulations. Although animal models of polyQ disease for understanding human pathology and accessing disease-modifying therapies in neurodegenerative diseases are available, there is neither a cure nor prevention for these diseases, and only symptomatic treatments for polyQ diseases currently exist. Long-term pharmacological treatment is so far disappointing, probably due to unwanted complications and decreasing drug efficacy. Cellular transplantation of stem cells may provide promising therapeutic avenues for restoration of the functions of degenerative and/or damaged neurons in polyQ diseases.
Keywords
Introduction
Polyglutamine (polyQ) diseases include spinocerebellar ataxias (SCA) types 1, 2, 6, 7 and 17, as well as Machado– Joseph disease (MJD/SCA3), Huntington's disease (HD), dentatorubral pallidoluysian atrophy (DRPLA), and spinal bulbar muscular atrophy X-linked type 1 (SMAX1/SBMA) (107,144). These diseases are autosomal dominantly inherited, except SMAX1/SBMA, which is linked to a mutation in the androgen receptor gene located on the X chromosome. The frequency of polyQ diseases averages 1–10 cases per 100,000 people (122). Of these polyQ disorders, HD and SCA3 have the highest prevalence worldwide (13). DRPLA predominantly occurs in Japan (198). SBMA has been reported with a high incidence in Finland (50,209). The cause of polyQ diseases is the expansion of trinucleotide cytosine–adenine–guanine (CAG) repeats encoding a polyQ tract in the coding region of causative genes. During protein synthesis, the expanded CAG repeats are translated into a series of uninterrupted glutamine residues forming a polyQ tract, and the accumulation of polyQ proteins may impair and damage the mitochondria, chaperone, and ubiquitin proteasome system (14,25,186). As a consequence, these aggregated polyQ proteins are found in the degenerated neurons, such as in the cerebellum, brainstem, and spinal tract (235). Therefore, different polyQ tract-containing proteins ultimately lead to the dysfunction and degeneration of specific neuronal subpopulations (57). The expanded CAG is unstable and tends to expand further, resulting in an earlier age of onset and a more severe disease course in successive generations of a kindred. That is a phenomenon named anticipation and is a prominent feature of all polyQ diseases (174). While polyQ diseases encompass nine different neurodegenerative disorders, each subtype of polyQ diseases has its own causative gene in different chromosomes and a threshold number of repeats (Table 1). These diseases also share some common pathological features, such as onset age at middle age; progressive worsening until death for 15–20 years; the longer the CAG repeat, the earlier the age of onset of the disease; the presence of mutant protein aggregates in selective degenerative neurons in specific regions of the brain. Several shared features of polyQ diseases suggest that the polyQ expansion may cause a common toxic effect in these different phenotypes.
Polyglutamine-Associated Diseases and Their Causative Genes and Proteins
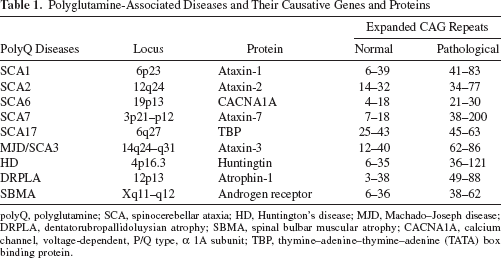
polyQ, polyglutamine; SCA, spinocerebellar ataxia; HD, Huntington's disease; MJD, Machado–Joseph disease; DRPLA, dentatorubropallidoluysian atrophy; SBMA, spinal bulbar muscular atrophy; CACNA1A, calcium channel, voltage-dependent, P/Q type, a 1A subunit; TBP, thymine–adenine–thymine–adenine (TATA) box binding protein.
Spinocerebellar Ataxia Types 1, 2, 3, 6, 7, and 17
Spinocerebellar ataxia (SCA), an autosominal dominant disorder, equally affects males and females and mainly attacks the central nervous system (CNS). Patients with SCA may show slowly progressive poor coordination of leg, hand, and eye movements as well as speech, leading to difficulties in walking, grasping, holding, etc. (165,174). Some specific types of SCA may be more aggressive, and most patients with SCA may require a wheelchair 10 to 15 years after the symptoms appear. Most patients with SCA have a shortened life span. The new subclassification system for SCA is numbered in chronological order of discovery of the gene locus and causes for each SCA type, now including SCA1 to 36, all of which are slightly different (165). For instance, SCA 8 is caused by a cytosine–thymine–guanine (CTG) repeat expansion, SCA10 is caused by an adenine–thymine–thymine–cytosine–thymine (ATTCT) repeat expansion, while SCA14 is not related to any repeat expansion at all. SCA14 is caused by a point mutation in the protein kinase C, γ (PRKCG) gene. A specific SCA subgroup, including types 1, 2, 3, 6, 7, and 17 (43), is of clinical importance for the unique gene mutations and phenotypes. In this review, we are focusing on this specific SCA subgroup.
Epidemiology
The prevalence of SCA estimates varies from 1–4/100,000 (120). The frequency of SCA has been assessed in different ethnic groups, including Taiwanese/Japanese/Caucasians in SCA1: 3%/3%/15%; SCA2: 0%/5%/14%; SCA3: 0.07%/0.11%/0.04%; SCA6: 18%/11%/5% (221); SCA7: 1.4% in Taiwan (76); SCA17: 0% (80,115).
Clinical Features
SCA1 is characterized by progressive loss of balance and coordination, impaired cognition, gaze palsy and slowing of saccades, dysarthria, dysphagia, peripheral neuropathy, pyramidal and extrapyramidal motor symptoms, executive dysfunctions, and respiratory failure (165). SCA1 usually starts in the mid-30s and progresses more rapidly than other SCA subtypes (120). Motor and sensory nerve conduction velocity (NCV) show a moderate slowing; visual-evoked potentials (VEPs) show a delayed P100 wave; somatosensory-evoked potentials (SSEPs) show loss of the P40 wave; brainstem auditory-evoked potentials (BAEP) show abnormalities; and motor-evoked potentials (MEPs) are lost (165).
SCA2 is characterized by progressive ataxia, dysarthria, posture tremor, slow saccades, hyporeflexia of the upper limbs, autonomic dysfunction, sleep disturbances, ophthalmoparesis, dementia, and parkinsonism (22). Onset age varies from 2 to 65 years, and onset before the age of 20 years correlates with a more aggressive disease course. NCV studies show an axonal sensory neuropathy; VEPs show a delayed or loss of the P100 wave; SSEPs and BAEPs are abnormal; and electrooculography (EOG) revealed severe slowing of saccades in gene carriers (165).
SCA3 is characterized by progressive cerebellar ataxia, areflexia, peripheral amyotrophy, muscle atrophy, parkinsonian features, dystonia, and spasticity (17). However, some minor presentations, such as external progressive ophthalmoplegia (EPO), dystonia, intention fasciculation-like movements of facial and lingual muscles, as well as bulging eyes, may also be of major importance for the clinical diagnosis of MJD (17). Age of onset varies from 5 to 75 years of age. SCA3 has several alternative names such as “Machado–Joseph disease,” “nigro-spino-dentatal degeneration with nuclear ophthalmoplegia,” “autosomal dominant striatonigral degeneration,” and “Azorean disease of the nervous system” (138,157,160,220). Presently, the most widely used designations are MJD and SCA3.
SCA6 is characterized by a slowly progressive ataxia, dysarthria, intention tremor, gaze-evoked and/or downbeat nystagmus, dysphagia, positional vertigo, and sensory, pyramidal, and extrapyramidal motor deficits (193). NCV studies show a mild axonal sensorimotor neuropathy; VEPs show a delay of the P100 wave; SSEPs show abnormalities; and BAEPs may be normal. EOG revealed an impaired smooth pursuit, dysmetric saccades, and impairment of vision-suppressed vestibule-ocular reflex (165).
SCA7, with onset in childhood, is characterized by macular or retinal degeneration with visual loss, slow saccades, ophthalmoplegia, progressive ataxia, dysphagia, and somatosensory and neuropsychiatric impairment (35). NCV studies show no significant findings; VEPs show loss of the P100 wave; and BAEPs may show abnormalities. EOG revealed visual dysfunction (165).
SCA17 is characterized by progressive gait and limb ataxia, seizure, cognitive dysfunctions, neuropsychiatric impairment, and pyramidal and extrapyramidal features such as spasticity, dystonia, chorea, and Parkinson's disease (105). NCV and VEPs are normal; BAEPs and SSEPs may show abnormalities (118).
Imaging
The findings of brain magnetic resonance imaging (MRI) in SCA include: SCA1 shows atrophy of the caudate nucleus, putamen, cerebellum, and brainstem; SCA2 shows substantial global atrophy of the cerebellum, thalamus, and brainstem (88); SCA3 shows enlargement of the fourth ventricle, moderate shrinkage of cerebellar vermis and hemispheres, and pontine, putaminal, and caudate atrophy (98). Positron emission tomography (PET) with fluorine-18-fluoro-2-deoxyglucose (FDG) showed significantly altered glucose metabolism in cerebellum, brainstem, and cerebral cortex in asymptomatic subjects with gene mutation, suggesting preclinical disease activity (188). In addition, these findings that included decreased FDG uptake in the cerebellar hemispheres, brainstem, and occipital cortex and increased FDG metabolism in the parietal and temporal cortices of asymptomatic SCA3 gene carriers are similar to those observed in Parkinson's disease. However, a controversial report, using [11C]d-threo-methylphenydate ([11C]dMP) and [18F]FDG to analyze the dopamine transporter and regional cerebral glucose metabolism (rCM-Rglu) in patients with SCA and Parkinson's disease, showed that the biochemical metabolism and pattern of dopamine terminal loss in SCA2 and SCA3 differed from that in Parkinson's disease (222). SCA6 shows atrophy in the cerebellar gray matter and alternations in cerebellar glucose metabolism (175). SCA7 shows atrophy of the cerebellum and brainstem. SCA17 shows a range of normal to moderate global atrophy or a focal atrophy of the cerebellum. In the chronic cases, the atrophy is prominent in the cerebellum, mild in the brain with sparing of the brainstem, and occasionally generalized cortical atrophy most obvious in the parietal lobe (34,62).
Molecular Genetics and Pathogenesis
Intranuclear inclusions containing the mutated protein are found in the cerebellar Purkinje cells and cortical neurons of affected individuals (156). Nuclear accumulated mutant proteins and inclusions have been identified as predominant in SCA1, SCA3, SCA7, and SCA17 patients. The underlying mechanisms of nuclear accumulation of expanded polyQ proteins in these polyQ diseases include gain-of-toxic effects, affecting gene expression, or disrupting nuclear organization and function. The numbers of uninterrupted CAG repeats on normal or patients' chromosome, mapping locations, and mutated proteins are listed in Table 1.
SCA1 is caused by an expansion of a CAG trinucleotide repeat that lies within the coding region of ataxin-1, which is predominantly nuclear in neurons and cytoplasmic in peripheral cells and involved in transcriptional regulation and RNA metabolism (126). Mice lacking ataxin-1 have spatial and motor learning deficiencies, and also impairment of short-term plasticity, but do not display ataxia or neuronal degeneration, suggesting the toxic gain-of-function mechanisms caused by the polyQ expansion (126).
SCA2 is caused by an expansion of a CAG trinucleotide repeat that lies within the coding region of ataxin-2, which is a cytoplasmic protein with highest expression in Purkinje cells (78). Cytoplasmic aggregation or accumulation of ataxin-2 has been shown to be sufficient to cause SCA2 pathology in humans and mice. Mutant ataxin-2 sensitizes Purkinje cells to glutamate-induced apoptosis. Glutamate-induced cell death of Purkinje cell cultures was attenuated by dantrolene, a clinically relevant ryanodine receptor inhibitor and Ca2 stabilizer, suggesting that the dantrolene may be a promising choice of treatment for SCA2 (115).
SCA3 is caused by an expansion of a CAG trinucleotide repeat that lies within the coding region of ataxin-2. Studies showed that in vitro cultured cells expressing ataxin-3 and expanded CAG activate apoptosis, and in vivo transgenic mice expressing the expanded polyglutamine stretch in Purkinje cells show ataxia, suggesting that the mutant protein is either directly or indirectly involved with a cellular suicide pathway, leading to ataxia (80). Ataxin-3 (ATXN3) interacts with UV excision repair protein Rad23 and valosin-containing protein (VCP), forming a protein complex. Rad23 and VCP are related to protein degradation machinery (40). Misfolding protein, caused by expanded CAG repeats, may lead to ubiquitination and subsequent formation of intranuclear inclusions (16). Parkin, the E3 ubiquitin ligase that is frequently mutated in early onset autosomal-recessive Parkinson's disease, promotes ubiquitination and degradation of ataxin-3 (206). The prion protein (PrP) promoter is employed in SCA3 models that express the full-length ATXN3 cDNA with various numbers of CAG repeats. The PrP/SCA3 homozygous mouse with ataxin-3 containing 71 glutamines shows posture abnormalities, muscle wasting, seizures, and progressive, ataxia-like motor dysfunction. Neuroanatomy shows no neuronal loss in the dentate nuclei but have a loss of tyrosine hydroxylase-positive neurons only in the substantia nigra (60). Mice expressing a 250-kb yeast artificial chromosome (YAC) construct that contains 50 kb of the human ATXN3 gene flanked by 30- and 170-kb genomic sequences show variety in the age of onset, mild to severe abnormal gaits, mild tremor, hypoactivity, and limb clasping (23).
SCA6 is caused by CAG repeat expansion in exon 47 at the 3′ region of the brain-specific, calcium channel, voltage-dependent, P/Q type, a 1A subunit (CACNA1A), which is highly expressed in Purkinje cells (234). When human CACNA1A gene with expanded CAG repeat is transfected into human embryo kidney (HEK) 293 cells, the voltage dependence of inactivation shifting negatively 6 to 11 mV was observed, suggesting a channelopathy. However, when CACNA1A protein and polyglutamine stretch were examined in SCA6 brain by immunohistochemistry, formations of aggregations within the cytoplasm of SCA6 Purkinje cells were seen (82–84,137), suggesting the causes of SCA6 are not only by channelopathy but also by a “gain-of-function.” Gabapentin, which interacts with the α2δ subunit of the P/Q-type voltage-dependent calcium channel, may be beneficial to SCA6 patients (137).
SCA7 is caused by expansion of an unstable trinucleotide CAG repeat encoding a polyglutamine tract in the corresponding protein, ataxin-7. Ataxin-7 is widely expressed in the cytoplasm and nucleus of nerve cells in regions that are either affected or unaffected by SCA7 pathology (75). Expression of the expanded ataxin-7 protein in transgenic mice leads to the development of intranuclear inclusions and the degeneration of rod photoreceptors and Purkinje cells, findings that are consistent with the human phenotype (106,230,232). SCA7 gene product, ataxin-7, is a subunit of the general control of amino acid synthesis protein 5 (GCN5) histone acetyltransferase-containing coactivator complexes, the thymine– adenine–thymine–adenine (TATA)-binding protein (TBP)-free TBP-associated factor-containing complex (TFTC), and the transcriptional adaptor 1 (suppressor of Ty insertions 3)/TBP-associated factor 9/GCN5 (SPT3/TAF9/GCN5) acetyltransferase complex (STAGA) (71). These transcriptional coactivator complexes can acetylate histone H3. The molecular pathogenesis may be caused by polyQ-expanded ataxin-7 deregulated TFTC/STAGA (70), suggesting a “transcriptionopathy.”
SCA17 is caused by a CAG/CAA repeat expansion of 45 or more in the TBP gene (101). TBP is the DNA-binding subunit of the RNA polymerase II transcription factor D (TFIID), which is essential for the expression of most protein-encoding genes (55,56), suggesting that polyQ expansion results in transcriptional dysregulation. Heat shock protein (HSP) 27 overexpression potentiates transcription factor, TATA-containing and TATA-lacking promoters, and specificity protein 1 (SP1). Coexpression of SP1 and HSP27 gene activates neurotrophic receptor genes low affinity nerve growth factor receptor [Ngfr(p75)] and high-affinity nerve growth receptor [tropomyosin-related kinase A (TrkA)]. Overexpression of HSP27 can rescue mutant TBP-mediated downregulation of TrkA, suggesting that nuclear HSP27 can promote neuronal protection in SCA 17 (54).
Pathology
SCA1 shows degeneration of the cerebellar Purkinje cells, brainstem cranial nerve nuclei, the inferior olive, and spinocerebellar tracts (97,98). SCA2 shows severe loss of Purkinje cells and of neurons in the substantia nigra and basis pontis. Mild to moderate neuronal loss in the inferior and accessory olives, dentate, arcuate, gracile, and accessory cuneate nuclei, internal granule cell layer of the cerebellum is also noted (78). The brain weight of SCA3 patients is significantly reduced, and neuronal loss in the cerebellar dentate nucleus, pons, substantia nigra, thalamus, globus pallidus, anterior horn cells, and Clarke's column in the spinal cord, vestibular nucleus, several cranial motor nuclei, and other brainstem nuclei occurs (43,59,100,164,194,223). Also, depigmented substantia nigra and atrophic changes of the cerebellum, pons, medulla oblongata, and the cranial nerves III to XII are reported in MJD brains, but the inferior olive and the cerebellar cortical neurons are typically spared (164,215,223). SCA6 demonstrates less severe degeneration in the cerebellum and neuronal loss with white matter involvement than other SCA families. The substantia nigra is also degenerative (58). SCA 7 shows progressive degeneration of the retina with loss of photoreceptors and damage to the retinal pigment epithelium. Neuronal loss is seen in the multiple grays of the cerebellothalamocortical and basal ganglia–thalamocortical loops of the somatomotor system and of the visual, auditory, and somatosensory systems. Myelin loss and/or atrophy is seen in the cerebellar white matter, corpus callosum, lateral lemniscus, oculomotor and abducens nerves, pontocerebellar and olivocerebellar fibers, trapezoid body, cuneate and gracile fascicles, internal arcuate fibers, and spinocerebellar tracts (166). SCA17 shows a mild global atrophy of the brain, predominant in the cerebellum because of severe Purkinje cell loss and Bergman's gliosis, but neuronal loss is mild in the dentate nucleus and the granular layer. Atrophy is moderate in the motor cortex and visual areas of the cerebrum. The pontine nuclei are spared, but the locus coeruleus and the substantia nigra are mildly affected (192).
Treatment
No treatment is currently available for SCA, and a quick and precise diagnostic method is necessary. Brain scans such as MRI and computerized tomography (CT) of affected persons' heads may show shrinkage of atrophy of the cerebellum, a typical finding of these diseases. In some cases, genetic testing may occasionally be useful when clinical presentations are suspected. However, there is variability in symptoms, severities, the rate of disease progression, and the onset age. All these may be obstacles to any asymptomatic subject with a positive genetic test for SCA screen. In fact, genetic tests may not always provide a clear diagnosis to most cases. Interestingly, the successful use of lithium (216) and neural stem cell transplantation in the cerebellum in SCA1 mouse models (32) may be applicable to patients with polyQ diseases.
Huntington's Disease
Huntington's disease (HD) is a rare, adult-onset, autosomal dominant, and progressive neurodegenerative disease (11,129,205). The diagnosis was historically based on the clinical picture of psychiatric, cognitive, and motor manifestations of the disease and the presence of a positive family history (124). HD at present can be confirmed by DNA determination because HD is known to be caused by an abnormal increase in CAG triplet repeats expansion in the huntingtin (htt) gene, which encodes an expanded polyQ stretch in the htt protein. Long polyQ tracts may possibly interfere with the normal function of cellular proteins, leading to the disease (128).
Epidemiology
The frequency of HD in Europe is 0.1–0.8/100000 (147,149,180); US and Canada: 0.3–0.69/100000 (2,103); China: 0.25–0.37/100000 (27); Taiwan: 0.08–0.42/100000 (30); and Japan: 0.65/100000 (139). Mean age at onset of symptoms is 30–50 years (8). In some cases, symptoms start before the age of 20 years with behavior disturbances and learning difficulties at school (30).
Clinical Features
The clinical diagnosis of HD is based on the development of chorea, which gradually spread to all muscles and is often observed in combination with other movement abnormalities, including dystonia, bradykinesia, and motor incoordination (214). Behavioral or psychiatric features, such as personality changes, poor attention, cognitive decline, irritability, and dementia are also common (158). The progression of the disease leads to a complete dependency in daily life, which results in patients requiring full-time care, and finally death approximately 15–20 years after disease onset (214). Owing to deterioration in the synchronizing movements, patients with HD may tend to have “drunk walk” and falls causing physical injuries (158). They may also have difficulty clearing the lungs and an increased risk of choking food or drink, leading to a risk of aspiration pneumonia (158). Suicide thoughts and committing suicides are not rare in these patients (214). The clinical assessment of the symptoms and signs of HD is important for patients, family, and caregivers. Currently, the diagnosis can be formally confirmed by DNA determination. Prenatal diagnosis is possible by chorionic villus sampling or amniocentesis (181).
Imaging
CT and MRI show caudate nuclei atrophy in the early course of HD and cerebral atrophy in the late stage. Functional MRI and PET can show changes in brain activity before the onset of symptoms (214).
Molecular Genetics and Pathogenesis
In HD, the expanded CAG repeats are located in the first exon of the htt gene, which codes huntingtin (HTT). The htt gene is located on chromosome 4p16. HTT is a 348-kDa multidomain protein that contains a polymorphic glutamine and proline-rich domain at its amino-terminus. The resulting mutant huntingtin protein with expanded CAG repeats may affect several cellular processes, such as transcriptional deregulation, mitochondrial dysfunction, and impaired vesicle transport (13,114,162). HTT also forms part of the dynactin complex, colocalizes with microtubules and interacting directly with β-tubulin, and involves clathrin-mediated endocytosis, suggesting a role in vesicle transport and/or cytoskeletal anchoring. Additionally, the longer polyQ domain seems to induce conformational changes of the HTT protein, which causes it to form intracellular aggregates that, in most cases, manifest as nuclear inclusion, in the nucleus, cell body, dendrites, and nerve terminals of neurons, and is also associated with a number of organelles including the Golgi apparatus, endoplasmic reticulum, and mitochondria (65,114). Mutant htt may possibly impair gene transcription, either by intranuclear aggregate formation or by sequestration of important transcription factors (24,65,114,195). Several transcription factors such as tumor protein 53 (p53), cAMP responsive element-binding protein (CREB)-binding protein (CBP), SP1, TBP, and molecular chaperones can be recruited to intranuclear aggregates (77,132,158,191). Also, the accumulation of proteasomes and ubiquitin in polyQ aggregates suggests that insufficient protein folding and degradation is implicated in the pathogenesis of polyQ disease (33,65,114,167). As a consequence of the presence of this expanded protein, the striatal medium-sized spiny neurons undergo selective degeneration accompanied in HD by progressive chorea and dementia (213), suggesting that huntingtin is important in the mature neurons of the brain during embryogenesis, and loss of huntingtin function may strongly contribute to the HD. Although most HD phenotypes always present late in life, the dominant mutation in the htt gene is present from birth. Moreover, the majority of HD patients have already had children before they are diagnosed, and they have thus passed the mutant htt gene on to the next generation (204). A gene test is necessary to any individual with a family history of HD. The gene test can detect the numbers of CAG repeats. In the unaffected population, the number of CAG repeats in the htt gene varies. CAG repeats in unaffected individuals range from 6 to 35; repeats of 36 or more define an HD allele. Individuals with 36–39 repeats are at increased risk for the development of HD. Individuals with 40 or more CAG repeats definitely manifest disease phenotypes (11,128,204). The longer the CAG repeat, the earlier the onset of disease.
Animal studies show that ablation of the htt gene in mice results in death at embryonic day 7.5 owing to aberrant brain development, and its conditional deletion in the forebrain also leads to neurodegeneration, suggesting that htt gene is necessary for cell survival and that its loss of function can be involved in neurodegeneration (44,140,233). Mutant huntingtin and the other polyQ disease proteins form insoluble aggregates in neurons. Abnormal accumulation of ubiquitinated proteins in the CNS is also involved in other neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease (202), suggesting that polyQ aggregates are toxic. A new technique, which can selectively inactivate wild-type huntingtin in animal brain, shows hypoactive and slight tremor at 10–12 months and a progressively more severe limbclasping-upon-tail suspension, a feature also observed in transgenic HD mice (74,119,151), suggesting that gene therapy and cell treatment approaches that influence the functions of htt gene may be practical and promising.
Dentatorubral Pallidoluysian Atrophy (DRPLA)
DRPLA, also known as Haw River syndrome and Naito–Oyanagi disease, is a rare autosomal dominant neurodegenerative disorder (20). Onset age ranges from 1 to 62 years with a mean age of onset of 30 years (81,104). The clinical manifestations are variable according to the onset age. The cardinal features in adults are ataxia, choreoathetosis, and dementia. Main features in children are progressive intellectual deterioration, behavioral changes, myoclonus, and epilepsy.
Epidemiology
DRPLA predominantly occurs in Japan (198) because more than 17 CAG repeats in ATN1 are significantly more frequent in the Japanese population than in populations of European origin. The prevalence rate of DRPLA in the Japanese population is estimated at 0.48/100,000 based on the nationwide study (207). Individuals with molecularly confirmed DRPLA have been found in other areas such as Europe and North America (109,125). However, no cases of DRPLA have been previously reported in Chinese (201,226) and Taiwanese (221) kindreds.
Clinical Features
There are at least three groups of DRPLA according to the onset age: the juvenile-onset group, with age less than 20 years, presents with ataxia and symptoms consistent with progressive myoclonus epilepsy; the early adult-onset group, with age between 20 and 40 years, may show seizures and myoclonus; and the late adult-onset group, with age more than 40 years, is characterized by ataxia, choreoathetosis, and dementia. Apart from these symptoms, cervical dystonia, corneal endothelial degeneration, autism, and surgery-resistant obstructive sleep apnea are not rare in patients with DRPLA (67).
Imaging
MRI of CNS may typically show atrophic changes in the cerebellum and brainstem, in particular the pontine tegmentum in the patients with DRPLA (110). Diffuse high signals in the white matter are often noted on T2-weighted MRI in the chronic cases with adult-onset DRPLA. Quantitative analyses revealed that the age and numbers of the expanded CAG repeat are related to the degree of atrophy of the brainstem and cerebellum, and the white matter changes (102).
Molecular Genetics and Pathogenesis
DRPLA is caused by a CAG repeat expansion in exon 5 of the ATN1 gene, which maps on chromosome 12p13.3 and encodes the 200-kDa atrophin-1 protein. Atrophin-1 is a known transcriptional coregulator, although its exact function is not well understood (170). In vitro studies have demonstrated that ATN1 is localized in both the nucleus and cytoplasm of neurons in the human CNS (224,227,228). The ATN1 gene encodes a hydrophilic 1,184-amino acid protein with several repetitive motifs including a serine-rich region, a variable length polyQ tract, a polyproline tract, and a region of alternating acidic and basic residues and a nuclear localizing signal (NLS) in the N-terminal and a nuclear export signal (NES) in the C-terminal domains. Cleavage of ATN1 to an N-terminal fragment relieves ATN1 of its nuclear export signal and concentrates it in the nucleus, leading to enhancement of cellular toxicity (142). The expanded CAG repeats create an adverse gain-of-function mutation in the gene products (228). The number of CAG repeats in the normal subject is 7–34, and in the affected individuals, it is with a repeat of 49 or more glutamines. The repeat increase in the expanded DRPLA gene may be more effective in jeopardizing neuronal cell than the changes in the Huntington disease and SCA1 genes (110).
The first mouse model of DRPLA is mice expressing full-length human ATN1 with 65 CAG repeats under transcriptional control of the mouse PrP showing progressive ataxia, tremors, aggression, and decreased breeding efficiency, and premature death (172). Similarly, nuclear accumulation was demonstrated, and occasional neuronal intranuclear inclusions were visualized, but no neuronal loss was seen in the brain (223). Neuronal intranuclear inclusions are immunopositive for several transcription factors (TF) such as TBP, TAF, CREB, and CBP (179,225). Recruitment of TFs into these neuronal intranuclear inclusions may induce transcriptional abnormalities, triggering progressive neuronal degeneration (223). Mice with ATN1 cDNA and 118 CAG repeats driven by a neuron-specific enolase (NSE) promoter show a similar, but a slightly more severe, phenotype of DRPLA than that of the PrP mice (229). The hemizygous transgenic offspring of the CAG 129 mice exhibited symptoms similar to juvenile-type DRPLA, such as ataxia, myoclonic movements, and tactile-induced and spontaneous epilepsy (170).
Pathology shows that these animals have severe brain atrophy manifesting as degeneration of dendrites and dendritic spines, presence of widespread neuronal intranuclear inclusions, and physiological changes in neurons, as recorded by electrophysiology (171). These abnormalities probably explain the brain atrophy and neuronal dysfunctions in this disease (168,170). In humans, DRPLA is characterized by marked, generalized brain and spinal cord atrophy, especially the dentatorubral and pallidoluysian systems (136,183). The pallidoluysian degeneration is more severe than the dentatorubral degeneration in juvenile onset; white matter changes were often detected in patients with onset of DRPLA in adulthood (79,148,223). Autopsy study of the white matter lesions showed diffuse myelin pallor, axonal preservation, and reactive astrogliosis in the cerebral white matter with only mild atherosclerotic changes (134). The diffuse accumulation of mutant ATN-1 proteins, found in the neurons and glial cells in the striatum, pontine, inferior olive, cerebellar cortex and dentate nucleus, is regarded as toxic (68). The extent and frequency of neurons showing the diffuse nuclear accumulations change depending on CAG repeat length. It is believed that the diffuse nuclear accumulations contribute to the clinical features such as dementia and epilepsy.
Spinal and Bulbar Muscular Atrophy (SBMA)
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease, was the first polyQ disease (94). SBMA is a rare, debilitating, neurodegenerative, X-linked, adult-onset motor neuron disease without effective treatment (50,94,95). The prevalence of this disease is about 1–2 per 100,000, whereas patients may have been misdiagnosed as having other neuromuscular diseases such as amyotrophic lateral sclerosis (51,61,145), but profound facial fasciculations, bulbar signs, gynecomastia, and sensory disturbances are the distinguished features from other motor neuron diseases. A high prevalence of SBMA has been reported in some areas like the Vasa region of Finland (50,209). The onset age is usually between 30 and 60 years of age, especially muscle weakness, but it is often preceded by nonspecific symptoms such as postural tremor and muscle cramps (7,63,72,190). Typically, affected individuals may require a wheelchair 15–20 years after the onset of weakness (7,26,72).
Clinical Features
Main symptoms of SBMA are weakness, atrophy, and fasciculations and are attributable to degeneration of lower motor neurons in the spinal cord and brainstem (64,95). Weakness and initial typical symptoms of SBMA are difficulty in walking and a tendency to fall. Bilateral facial and masseter muscle weakness, poor uvula and soft palatal movements, and atrophy of the tongue with fasciculations are often encountered. Fasciculations are not prominent at rest, but apparent on voluntary muscle movement. These contraction fasciculations are obvious in the face, neck, tongue, and limbs (49). In extremities, involvement is usually predominant in proximal musculature and is occasionally asymmetric (7,113). Decreased or absent deep tendon reflexes without pathological reflex are noted (4,49). Sensory involvement is largely restricted to a sense of vibration, which is affected distally in the legs (185). Muscle tone is usually hypotonic, and no pyramidal signs are detected. Cerebellar symptoms are absent, while dysautonomia and mild cognitive impairment have been reported in a limited number of patients (131,155). Neuromuscular symptoms are often worsened by coldness and by fatigue after exercise. Patients occasionally experience laryngospasm, a sudden sensation of dyspnea (189). Advanced cases often develop dysphagia, eventually resulting in aspiration or choking. Reduced velopharyngeal closure may result in speech with a nasal component (49). EMG shows neurogenic abnormalities, and NCV shows prolonged distal motor latencies (72,196). Patients often show symptoms related to androgen insensitivity, such as gynecomastia, testicular atrophy, dyserection, and decreased fertility (12,36,190). Abdominal obesity is common, whereas male pattern baldness is rare in patients with SBMA (182). The vast majority of patients with SBMA have a normal life expectancy and do not die from direct complications of their disease.
Imaging
MRI of the brain shows loss in the white matter in multiple areas, including cerebellar hemispheres, the tectum of the midbrain, and the medial lemniscus in the dorsal brainstem. In addition, atrophy was localized both in the subcortical areas of the temporal and the occipital lobe and in the parietal lobe. However, the atrophic levels of gray matter or white matter did not correlate with CAG repeat number (89). MRI shows significant alterations in the metabolite ratios in parts of the brainstem and motor regions of the brain, suggesting this disease may be due to metabolites and genetic failure (117).
Molecular Genetics and Pathogenesis
The principal mutation causing SBMA is an abnormal expansion of CAG repeats in exon 1 of the androgen receptor gene, and loss of motor neurons in the spinal cord and brainstem are associated with them (107). The androgen receptor, which gene is located on Xq11–q12, is a nuclear receptor that normally regulates gene expression after ligand binding. Similar to other triplet repeat diseases, the age at onset of the disease has been inversely linked to the numbers of CAG-repeat expansions (161,169). In unaffected individuals, this region ranges in size from 9 to 34 repeats, whereas individuals affected with SBMA have 36–66 repeats. However, some SBMA studies reported no correlation between the progression of the clinical course and the number of CAG repeats (116,190), but a positive correlation between ages and severity of limb muscle weakness (41). The length of the CAG expansion correlates inversely with age of disease onset, that is, the longer the expansion, the earlier the onset (108). CAG repeat length has also been reported to correlate with motor and sensory nerve conduction abnormalities (196). The primary androgen receptor ligands are testosterone and dihydrotestosterone. Animal models show male transgenic mice expressing the human AR gene with expanded CAG repeats showing progressive muscular atrophy and weakness associated with the nuclear accumulation of mutant AR protein similar to SBMA. When these male mice are castrated (31) or treated with the antiandrogen (90,91), these deficits are improved (173,184). Additionally, the pathogenic AR is found to inhibit the transcription of transforming growth factor-β receptor type II (TGF-βRII) via abnormal interactions with nuclear transcription factor Y (NF-Y) and p300/CBP-associated factor, enhancing polyQ-induced cytotoxicity in an in vitro cell study (92), suggesting that these ligands are important in the development of the disease, and TGF-bRII may be one of the important effectors for these ligands. Moreover, the clinical and pathological phenotypes of these mice were markedly improved by the overexpression of heat shock pro teins (1,93), and a potent HSP90 inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG) (217), can improve motor function deficits and pathological changes in SBMA transgenic mice. These remarkable findings support the potential therapeutic effects of these approaches in patients with SBMA.
Diagnosis and Treatment
Diagnosis of polyQ diseases relies on positive family history, clinical findings, and genetic tests. For symptom treatment, levodopa is temporarily useful for rigidity/bradykinesia and for tremor; magnesium for muscle cramp; baclofen for spasticity; prism glasses for diplopic symptoms; and rehabilitation programs, containing physiotherapy and/or physical therapy, for gait instabilities, maintaining the mobility of joints, prevention from falls, and strengthening patients' independence. Walkers and wheelchairs can greatly help the patient with everyday tasks. These treatments provide only partial and temporary alleviation of symptoms.
Gene Therapy and Stem Cell Transplant
Gene Therapy
Delivering gene(s) to reduce mutant protein levels leading to a decrease in all downstream toxic effects can function by using antiaggregate chemicals (200,234), intracellular antibodies (19,21), activation of the ubiquitin–proteasome system (135,176,199), or inducing auto phagic pathway (73,85,129,130,159) genes. Knockdown gene by RNAi, short hairpin RNA (shRNA), short interfering RNA (siRNA), and single-stranded antisense oligonucleotides (AONs) can inhibit the formation of mutant polyQ proteins (39,42). AONs show a significant reduction in expanded transcript levels in patient-derived fibroblasts from SCA1, SCA3, DRPLA, and HD (48), supporting that one single antisense oligonucleotide may be a potential therapeutic option for polyQ diseases. However, the expression of a CAG repeat transcript from the antisense strand produces a toxic polyQ protein in a mouse model (219). There are a number of viral vectors that can be used for gene delivery to the CNS. Two main viral vector systems are lentiviruses (LV) and adeno-associated viruses (AAV). Both viruses are minimally immunogenic and can transduce a number of CNS cell types. There are several differences between LV and AAV. One is that most LV vectors integrate, while AAVs do not. The other is the level of expression because LV-mediated transduction often results in low copy numbers of transgene/cell. Last, but not the least, is that the placement of the expression cassette in the LV genome can affect expression levels (66). However, both of them have been used successfully in both gain-of-function studies (37) and loss-of-function studies (42,150). Therefore, gene interventions may have potential and are practical for treating these incurable diseases in the future.
Stem Cell Transplantation
Using fetal striatal cells or medium spiny neurons derived from embryonic stem cells in animal models and patients showed significant improvement (15,28,153), indicating that grafted fetal neurons can provide therapeutic benefit in patients with polyQ diseases. However, transplanted fetal striatal cells become necrotic or apoptotic within the first 24 h (46), and it is therefore difficult to obtain sufficient amounts of embryonic striatal tissue as well as the concomitant ethical issues associated with the use of human embryonic tissue (15,18,236). Stem cells can provide an extensive source for maintaining cell numbers from the growing, injured, diseased cells. Grafting murine neural precursor cells promoted cerebellar Purkinje cell survival and functional recovery in an SCA1 mouse model (32), and transplanting murine mesenchymal stem cells to a mouse cerebellum in an animal model of cerebellar ataxia improved motor functions and rescued degenerated Purkinje cells (87). These findings support the idea that stem cell transplantation may be applicable in patients with incurable polyQ diseases. There are several types of stem cells, including embryonic stem cells, neural stem cells, mesenchymal stem cells, and induced pluripotent stem cells, which will be discussed below.
Embryonic Stem Cells (ESCs)
ESCs are isolated and expanded from the inner cell mass of the mammalian blastocyst stage embryo. ESCs can proliferate extensively in vitro and can produce every cell type of the body, including neurons, astrocytes, and oligodendrocytes (10,143). In this way, human ESCs could be considered the best source for cell transplantation. A number of human lines have now been generated (152,205), and in some countries, banking of such cells is being developed. Human ESCs can be induced to differentiate to neurons by using a variety of methods (96,218). However, the lack of genetic stability and long-term survival, risk of tumor formation, and the need of destroying a human embryo to obtain hESC lines, as well as the possible and potential carcinogenesis, limit research for the use of these cells.
Neural Stem Cells (NSCs)
NSCs, identified in the subventricular zone of humans, are the earliest uncommitted multipotent cells in the CNS (5). NSCs have the abilities of extensive proliferation and self-renewal. They can differentiate into neurons, oligodendrocytes, and astrocytes, and participate in normal CNS development and differentiate into regionally appropriate cell types in response to environmental factors (47,53,163). NSCs can be isolated from almost all parts of the embryonic/fetal brains and can be propagated in vitro and subsequently implanted into the brain of animal models of human neurological disorders. However, NSCs in different regions of brains may possess variable proliferative, self-renewal, and differentiation abilities. NSCs are produced by placing dissociated fetal brain cells into serum-free medium with mitogens such as fibroblast growth factor 2 (FGF-2) and epidermal growth factor (EGF). Under these conditions, differentiated cells die, and more primitive cells proliferate as free-floating spheres of cells, the so-called “neurospheres” (154). Continuously dividing immortalized human NSCs with switch-on oncogenes have advantageous features including homogeneous nature and the same clone (6,154). It requires significant advances in our understanding of the developmental signals for proliferation, differentiation, and integration of NSCs and to determine favorable conditions of the host brain environment for implanted NSCs to survive, prosper, and restore the damaged brain by stem cell transplant therapy.
Mesenchymal Stem Cells (MSCs)
Bone marrow contains hematopoietic and MSCs, which are defined as plate-adhering, fibroblast-like cells possessing self-renewal ability with the capacity to differentiate into multiple mesenchymal cell lineages such as osteoblasts, chondrocytes, and adipocytes. MSCs are easily accessible and isolated from a variety of tissues such as adipose tissue, umbilical cord and umbilical cord blood, placental tissue, liver, spleen, testes, menstrual blood, amniotic fluid, pancreas and periosteum, and infant dental pulp (210). Under appropriate conditions, MSCs can differentiate into skeletal, cardiac muscle, lung, and liver cells and can migrate toward damaged tissues in an animal model of strokes and Parkinson's disease (29,69). MSCs also provide the advantage of minimizing immune reactions because cells can be derived from the respective patient. Furthermore, several human trials of MSCs have shown no adverse reactions to allogenic MSC transplants (52,123). Although the mechanisms are not clear, chemokine (C-X-C motif) receptor 4 (CXCR4), expressed on MSCs, can be navigated by a signal molecule, stromal cell-derived factor-1α (SDF-1α), which may direct MSCs to ischemic and degenerative lesions to repair injured tissues in the brain (9,121). Studies have shown that rat and human MSCs can differentiate into neuron-like cells and can enter mouse CNS (121,197). All these data suggested that cell therapy with MSCs for polyQ diseases may be feasible, easy, and most importantly there are no ethical problems. Transgenic animal studies show intravenous transplantation of hMSCs can indeed delay the onset as well as improve the motor function of SCA2 transgenic mice. Therefore, MSC transplantation trials in multiple system atrophy (112), Parkinson's disease (211), amyotrophic lateral sclerosis (127), and ischemic stroke (111) are with promising results.
Induced Pluripotent Stem Cells (iPSCs)
iPSCs are generated from skin fibroblasts through the activation of a combination of genes that are capable of dedifferentiating the fibroblasts into a pluripotent cell (231) and neural lineages (212). iPSCs can be generated from animals, humans, and patients with diseases, such as amyotrophic lateral sclerosis (38), spinal muscular atrophy (45), and Parkinson's disease (187), polyQ diseases including HD, SCA3, and SBMA (99,141,146,177). Therefore, human iPSCs have emerged as a useful tool for research into the pathogenesis of many diseases and developing new therapeutic strategies. There are two studies that reported the generation and characterization of iPSC-derived models for HD and the genetic correction of a disease-causing CAG repeat expansion mutation in iPSCs from individuals with HD (3,203), supporting that the transplantation of iPSC-derived neurons with normal or better functional activity to patients with polyQ diseases is going to become an option of total cure.
Others
Umbilical cord blood (UCB) is another source of hematopoietic and nonhematopoietic stem cells. Human UCB (hUCB) stem cells can be induced to differentiate into neuron-like cells, astrocytes, and oligodendrocytes by the treatment of retinoic acid (RA) (86). Human embryonic germ (hEG) cells, derived from primordial germ cells in the gonadal ridge of first-trimester embryos (178,208), can replace the damaged neurons and oligodendrocytes in the forebrain of neonatal mice (133), and their neural progenitors are capable of robust and long-term growth. However, the low efficiency, limited proliferation rate, and difficult control of differentiation may restrain the use of hEG cells (178). Although there are still numerous obstacles to overcome before clinical application of cell therapy in patients with these degenerative neurological diseases, a Phase I clinical trial confirmed that MSC transplantation into the spinal cord of ALS patients is safe (127), suggesting that, in theory, stem cell therapy can indeed restore or preserve brain function by replacing and protecting neurons, but it is still uncertain which stem cells are most suitable as cellular grafts. Also, it is not clear how and what factors fully control the differentiation, survival, and maturation of stem cells in the recipient's degenerative brain. A well-designed protocol, clearly indicating which stem cells and growth factors and timing is necessary for effective cell transplant therapy for treating degenerative diseases.
Footnotes
Acknowledgment
Supported by Industry-Academy Cooperation Office (10142668) and MOHW103-TD-B-111-03. The authors declare no conflicts of interest.